
Flu Shot
Introduction
In this section we will:
* identify which Flu Shot procedures were excluded and why
* identify which different Flu Shot Replacement subgroups were created and why
If you have questions or concerns about this data please contact Alexander Nielson (alexnielson@utah.gov)
Load Libraries
Load Libraries
library(data.table)
library(tidyverse)## Warning: replacing previous import 'vctrs::data_frame' by 'tibble::data_frame'
## when loading 'dplyr'library(stringi)
library(ggridges)
library(broom)
library(disk.frame)
library(RecordLinkage)
library(googlesheets4)
library(bigrquery)
library(DBI)
devtools::install_github("utah-osa/hcctools2", upgrade="never" )
library(hcctools2)Establish color palettes
cust_color_pal1 <- c(
"Anesthesia" = "#f3476f",
"Facility" = "#e86a33",
"Medicine Proc" = "#e0a426",
"Pathology" = "#77bf45",
"Radiology" = "#617ff7",
"Surgery" = "#a974ee"
)
cust_color_pal2 <- c(
"TRUE" = "#617ff7",
"FALSE" = "#e0a426"
)
cust_color_pal3 <- c(
"above avg" = "#f3476f",
"avg" = "#77bf45",
"below avg" = "#a974ee"
)
fac_ref_regex <- "(UTAH)|(IHC)|(HOSP)|(HOSPITAL)|(CLINIC)|(ANESTH)|(SCOPY)|(SURG)|(LLC)|(ASSOC)|(MEDIC)|(CENTER)|(ASSOCIATES)|(U OF U)|(HEALTH)|(OLOGY)|(OSCOPY)|(FAMILY)|(VAMC)|(SLC)|(SALT LAKE)|(CITY)|(PROVO)|(OGDEN)|(ENDO )|( VALLEY)|( REGIONAL)|( CTR)|(GRANITE)|( INSTITUTE)|(INSTACARE)|(WASATCH)|(COUNTY)|(PEDIATRIC)|(CORP)|(CENTRAL)|(URGENT)|(CARE)|(UNIV)|(ODYSSEY)|(MOUNTAINSTAR)|( ORTHOPEDIC)|(INSTITUT)|(PARTNERSHIP)|(PHYSICIAN)|(CASTLEVIEW)|(CONSULTING)|(MAGEMENT)|(PRACTICE)|(EMERGENCY)|(SPECIALISTS)|(DIVISION)|(GUT WHISPERER)|(INTERMOUNTAIN)|(OBGYN)"Connect to GCP database
bigrquery::bq_auth(path = 'D:/gcp_keys/healthcare-compare-prod-95b3b7349c32.json')
# set my project ID and dataset name
project_id <- 'healthcare-compare-prod'
dataset_name <- 'healthcare_compare'
con <- dbConnect(
bigrquery::bigquery(),
project = project_id,
dataset = dataset_name,
billing = project_id
)Get NPI table
query <- paste0("SELECT npi, clean_name, osa_group, osa_class, osa_specialization
FROM `healthcare-compare-prod.healthcare_compare.npi_full`")
#bq_project_query(billing, query) # uncomment to determine billing price for above query.
npi_full <- dbGetQuery(con, query) %>%
data.table() get a subset of the NPI providers based upon taxonomy groups
gs4_auth(email="alexnielson@utah.gov")surgery <- read_sheet("https://docs.google.com/spreadsheets/d/1GY8lKwUJuPHtpUl4EOw9eriLUDG9KkNWrMbaSnA5hOU/edit#gid=0",
sheet="major_surgery") %>% as.data.table## Auto-refreshing stale OAuth token.## Reading from "Doctor Types to Keep"## Range "'major_surgery'"surgery <- surgery[is.na(Remove) ] %>% .[["NUCC Classification"]] npi_prov_pair <- npi_full[osa_class %in% surgery] %>%
.[,.(npi=npi,
clean_name = clean_name
)
] Load Data
bun_proc <- disk.frame("full_apcd.df")flu <- bun_proc[proc_code_str_sorted %>% stri_detect_regex("90(672|686|688|682|685|687|685|662|653|674|756)")]flu <- flu[tp_med < 1500 & cnt > 5 & surg_bun_sum_med == 0 & radi_bun_sum_med==0 & anes_bun_sum_med==0 & duration_mean == 0 ]flu %>% plot_med_density() %>% print()## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.
flu %>% get_tag_cor() %>% print()## Warning in stats::cor(cor_data): the standard deviation is zero## # A tibble: 329 x 2
## name correlation
## <chr> <dbl>
## 1 medi_bun_t_pcv13 0.493
## 2 medi_bun_t_dtap 0.426
## 3 faci_bun_t_est 0.358
## 4 medi_bun_t_oral 0.341
## 5 medi_bun_t_dtap-ipv 0.340
## 6 medi_bun_t_mmr 0.310
## 7 medi_bun_t_hepb 0.309
## 8 medi_bun_t_hpv 0.271
## 9 medi_bun_t_menacwy 0.255
## 10 medi_bun_t_immun 0.237
## # ... with 319 more rowsflu + Pneumococcal conjugate vaccine
flu %>% get_tag_density_information(tag="medi_bun_t_pcv13") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ medi_bun_t_pcv13"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 18.8## $dist_plots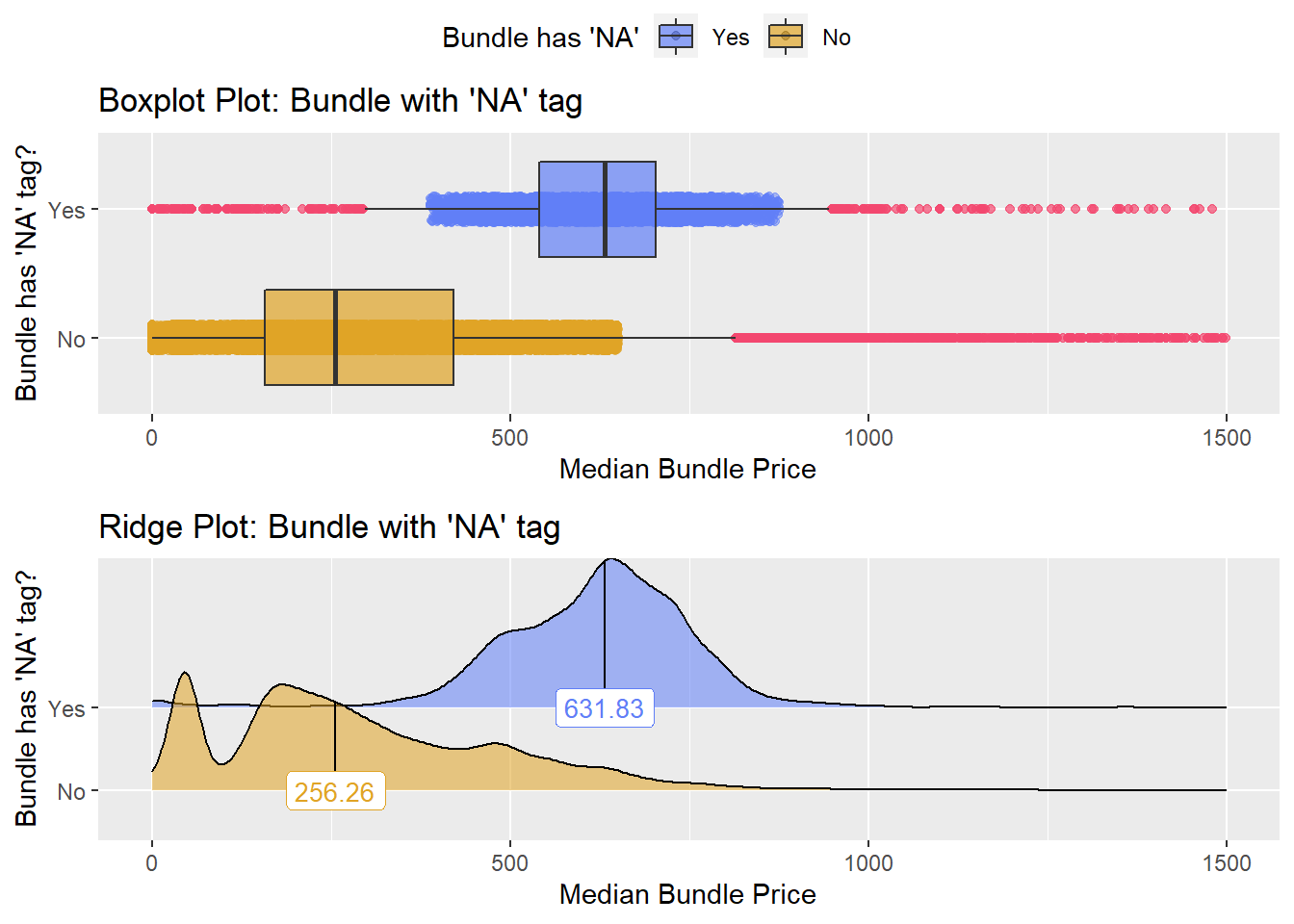
##
## $stat_tables
flu_w_pcv13 <- flu[medi_bun_t_pcv13==T]
flu <- flu[medi_bun_t_pcv13==F]dtap
flu %>% get_tag_density_information(tag="medi_bun_t_dtap") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ medi_bun_t_dtap"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 20.8## $dist_plots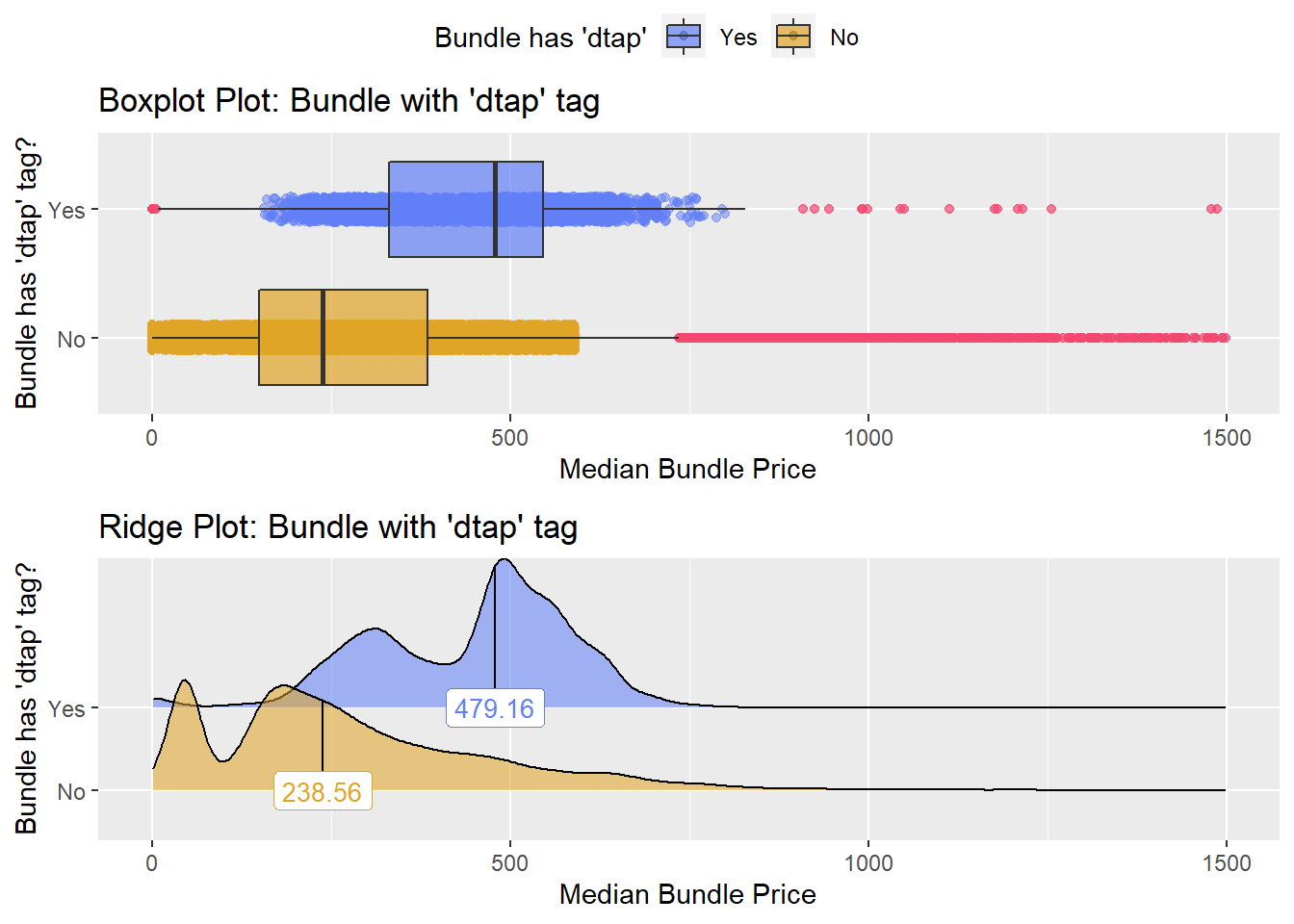
##
## $stat_tables
flu_w_dtap <- flu[medi_bun_t_dtap==T]
flu <- flu[medi_bun_t_dtap==F]flu with hepb
flu %>% get_tag_density_information(tag="medi_bun_t_hepb") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ medi_bun_t_hepb"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 24.9## $dist_plots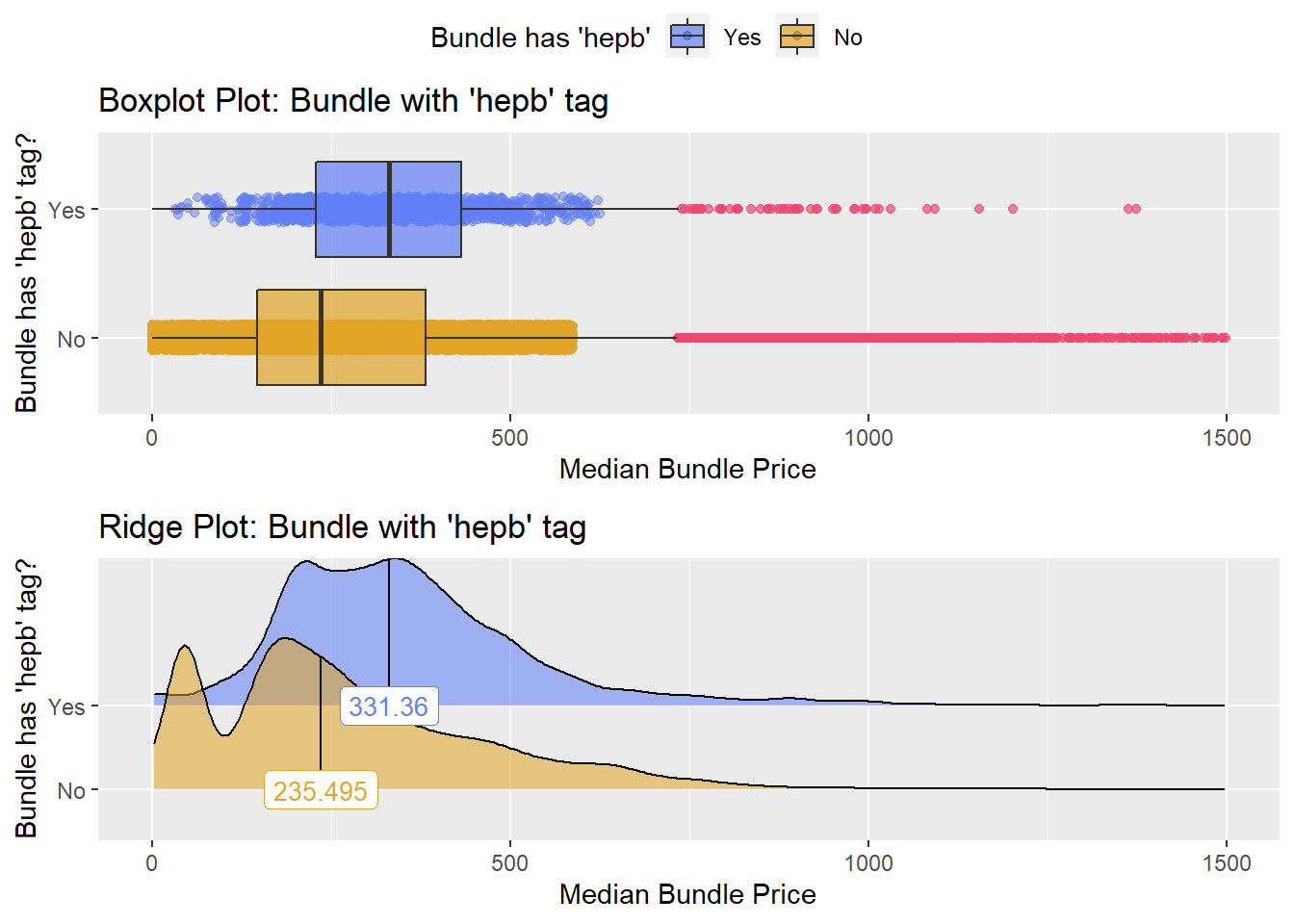
##
## $stat_tables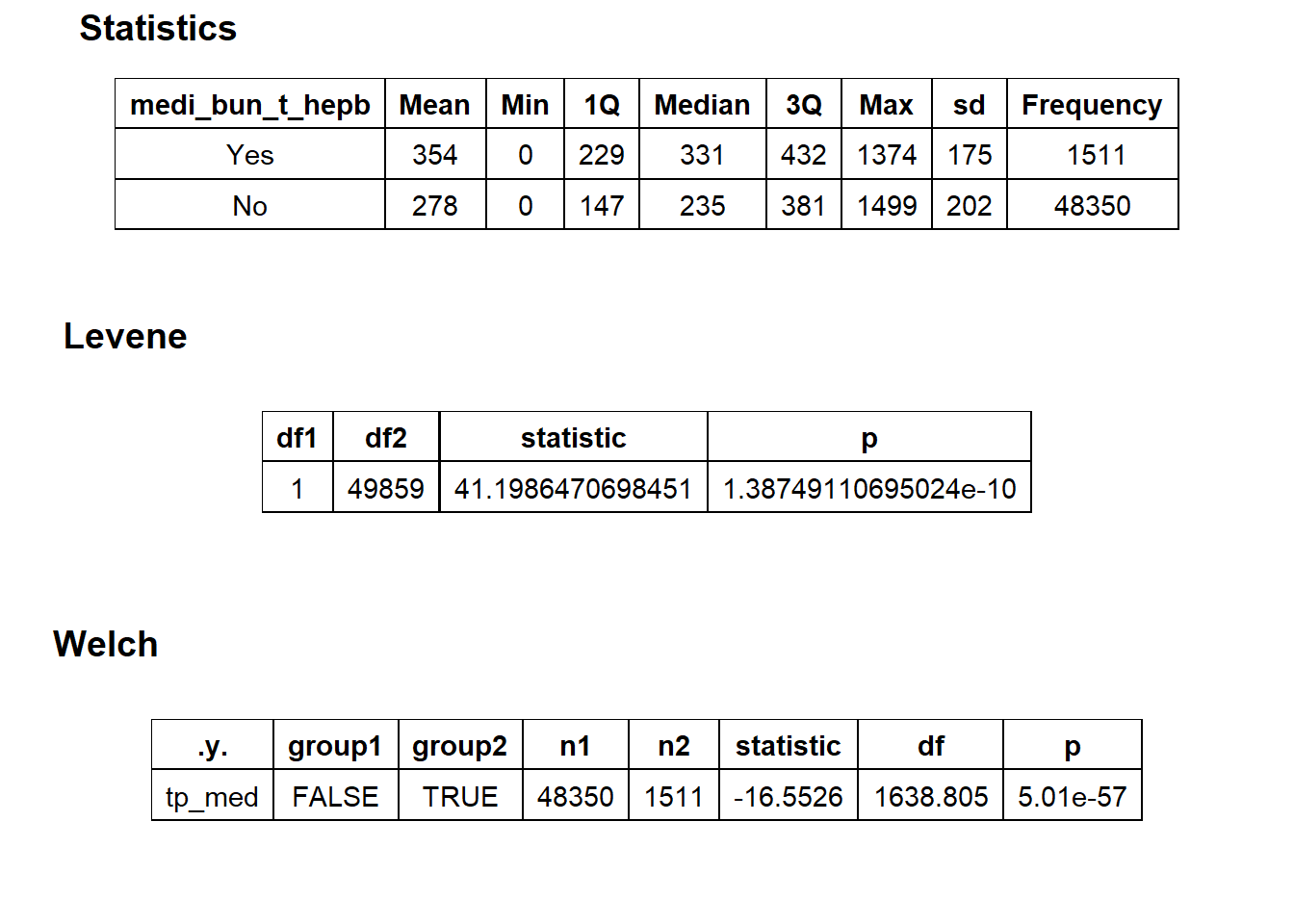
flu_w_hepb <- flu[medi_bun_t_hepb==T]
flu <- flu[medi_bun_t_hepb==F]hpv
flu %>% get_tag_density_information(tag="medi_bun_t_hpv") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ medi_bun_t_hpv"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 20.8## $dist_plots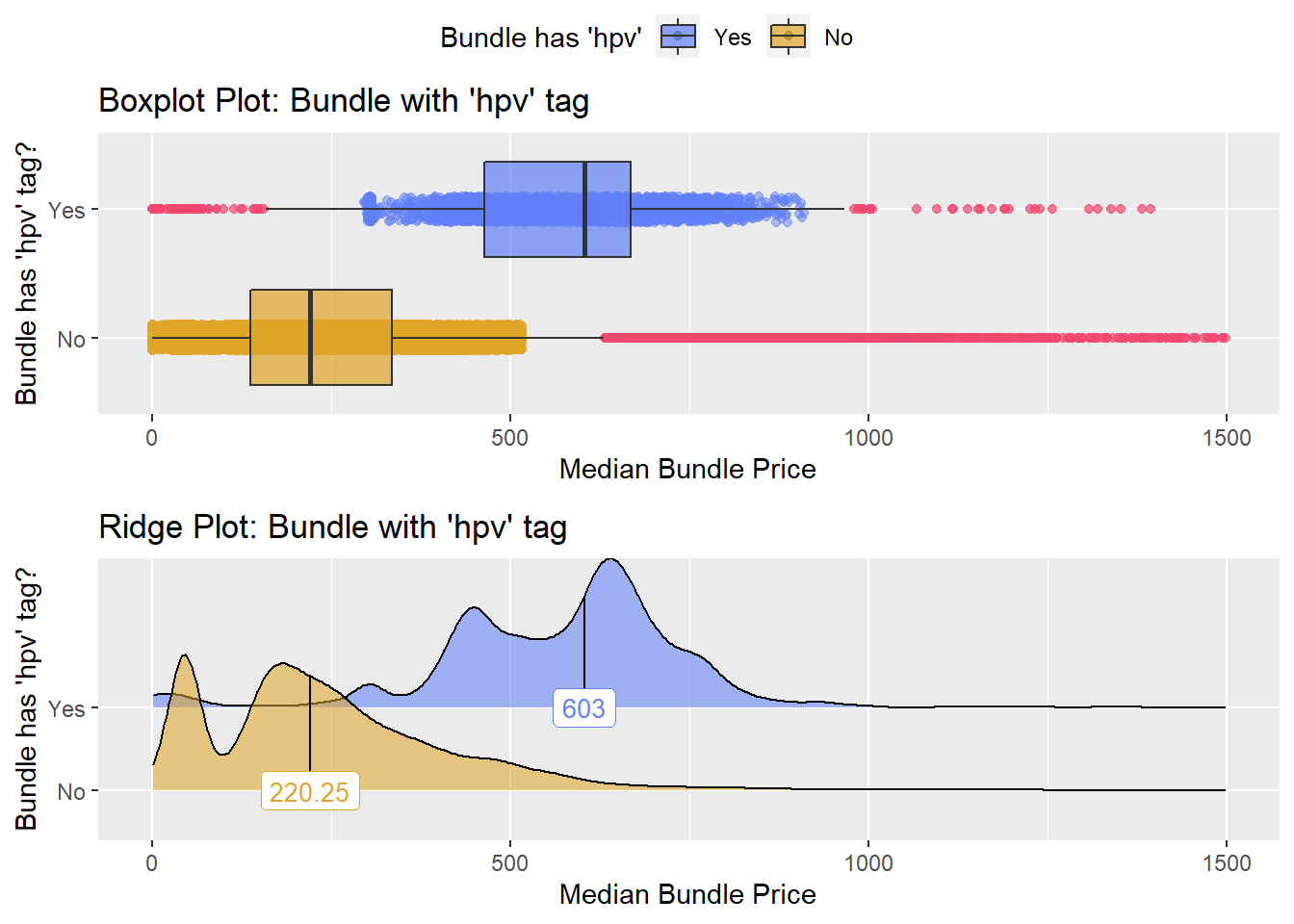
##
## $stat_tables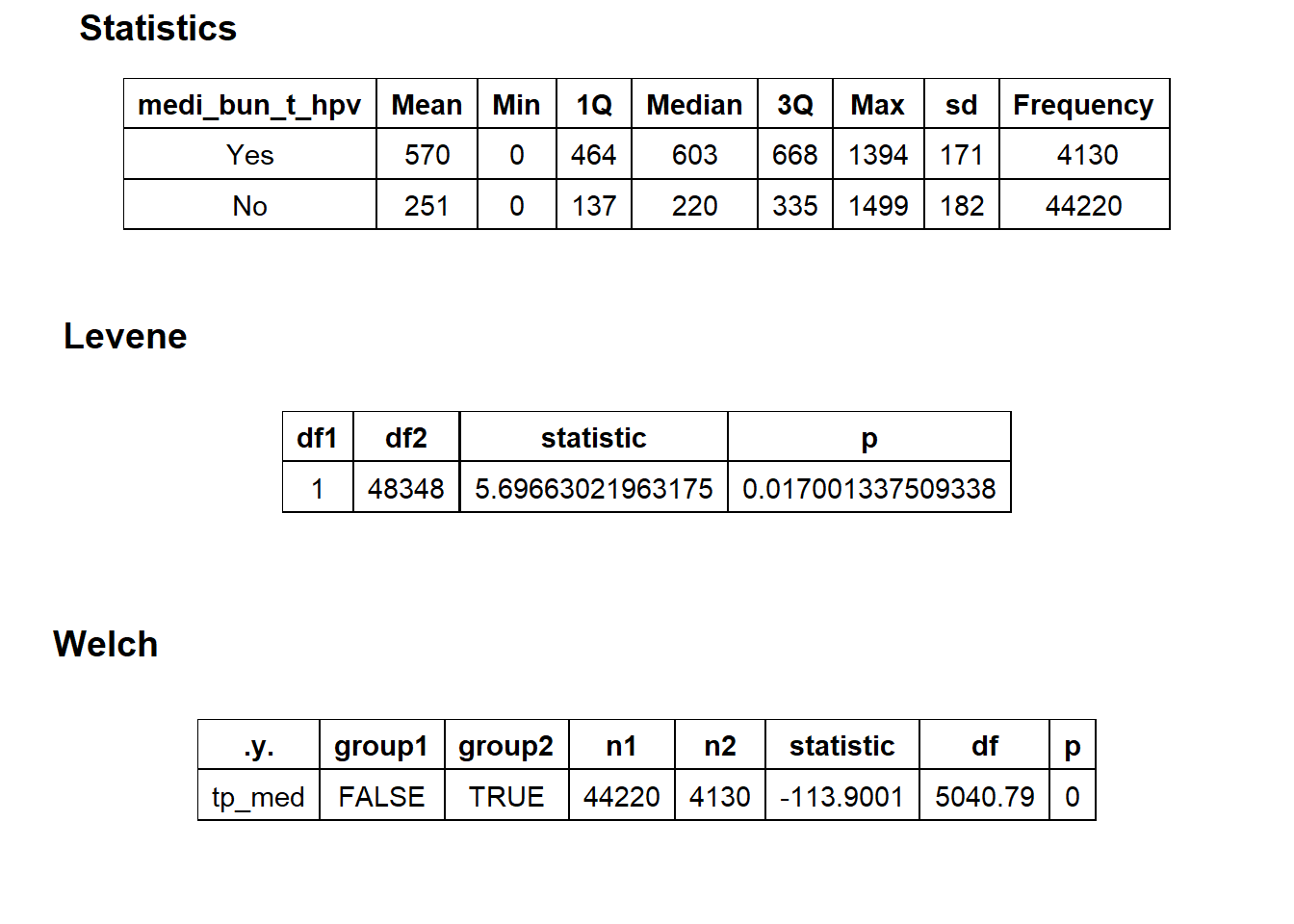
flu_w_hpv <- flu[medi_bun_t_hpv==T]
flu <- flu[medi_bun_t_hpv==F]menacwy
flu %>% get_tag_density_information(tag="medi_bun_t_menacwy") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ medi_bun_t_menacwy"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 19.8## $dist_plots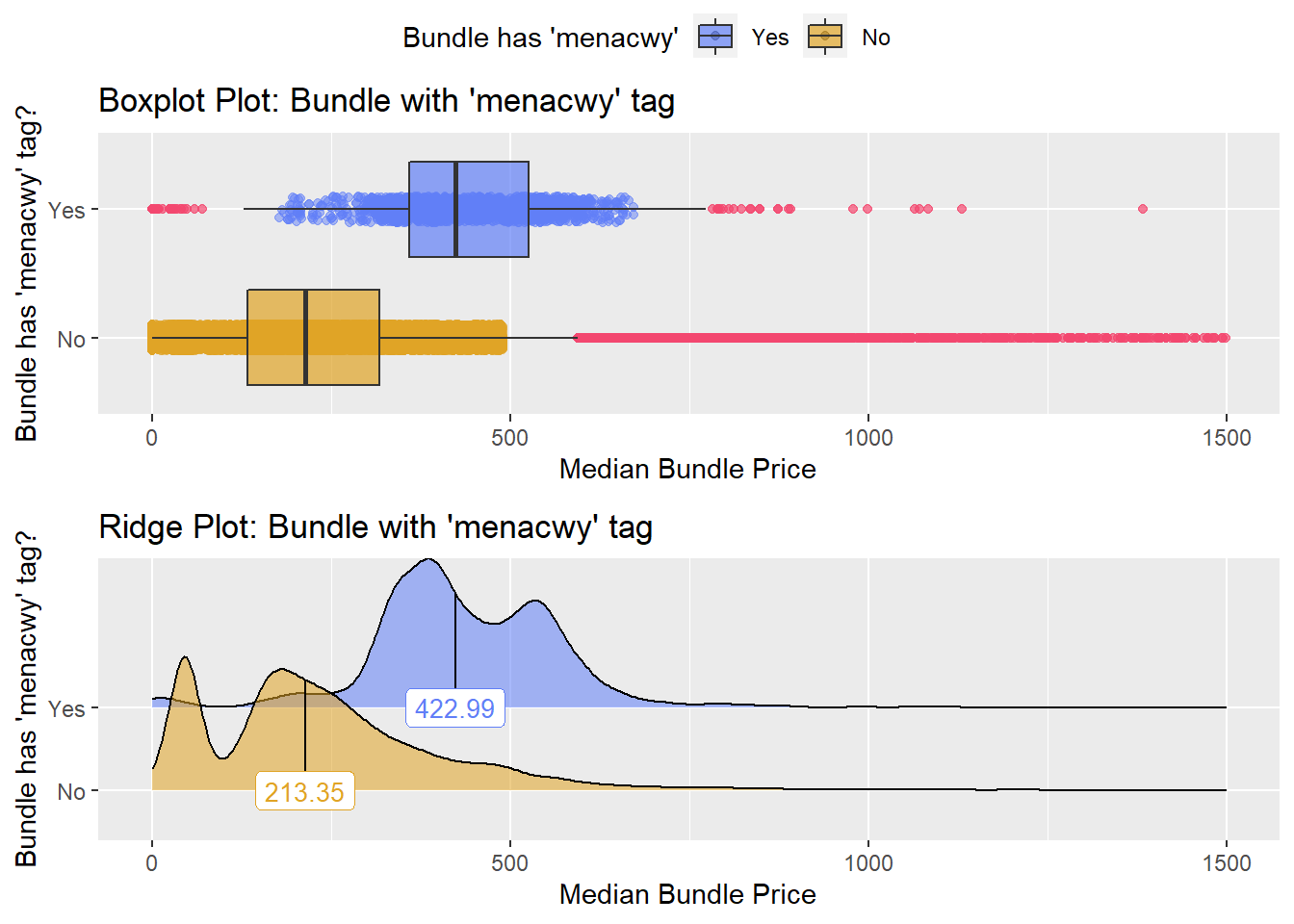
##
## $stat_tables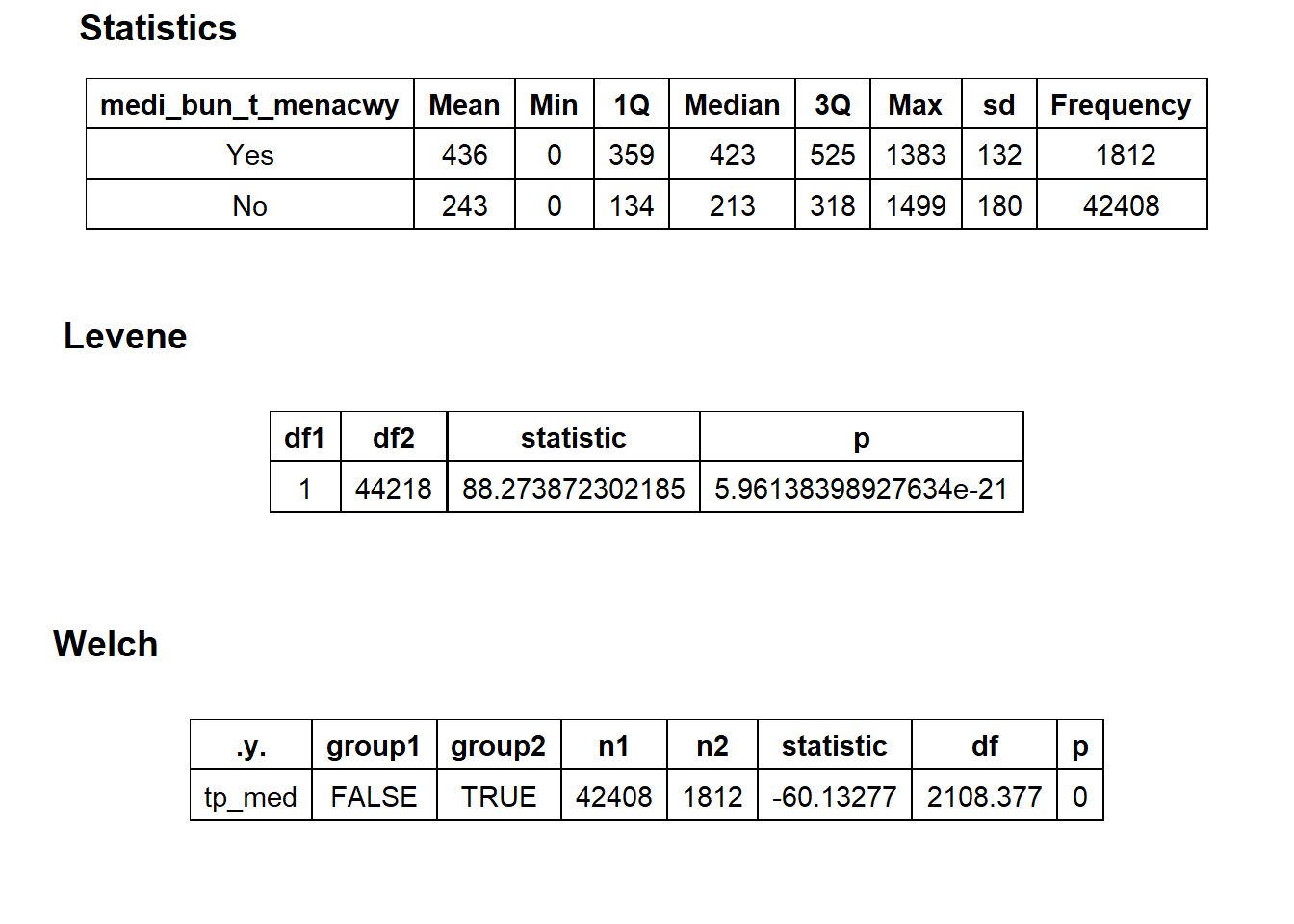
flu_w_menacwy<- flu[medi_bun_t_menacwy==T]
flu <- flu[medi_bun_t_menacwy==F]flu %>% get_tag_cor() %>% print()## Warning in stats::cor(cor_data): the standard deviation is zero## # A tibble: 315 x 2
## name correlation
## <chr> <dbl>
## 1 faci_bun_t_visit 0.487
## 2 path_bun_t_panel 0.392
## 3 faci_bun_t_est 0.359
## 4 path_bun_t_assay 0.357
## 5 path_bun_t_lipid 0.304
## 6 path_bun_t_test 0.278
## 7 path_bun_t_metabolic 0.267
## 8 path_bun_t_hemoglobin 0.262
## 9 medi_bun_t_mmr 0.251
## 10 path_bun_t_comprehen 0.251
## # ... with 305 more rowsflu <- flu[path_bun_t_assay==F & path_bun_t_lipid==F & path_bun_t_metabolic==F & path_bun_t_hemoglobin==F & path_bun_t_comprehen==F & path_bun_t_complete==F &
path_bun_t_vitamin==F & path_bun_t_hormone==F & path_bun_t_thyroid==F &
path_bun_t_hep==F]flu %>% plot_med_density() %>% print()## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.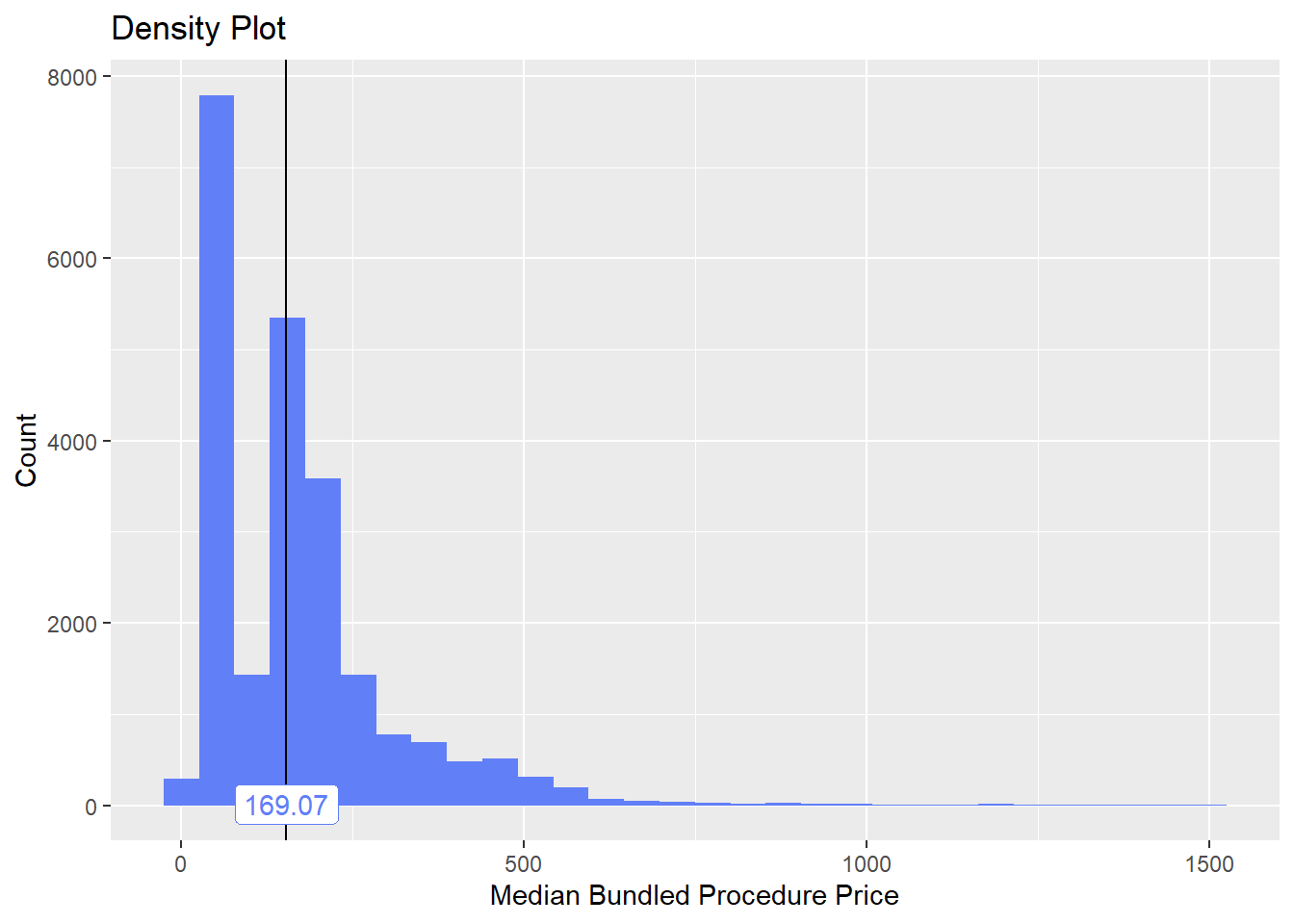
flu %>% get_tag_cor() %>% print()## Warning in stats::cor(cor_data): the standard deviation is zero## # A tibble: 229 x 2
## name correlation
## <chr> <dbl>
## 1 faci_bun_t_visit 0.531
## 2 faci_bun_t_est 0.458
## 3 medi_bun_t_mmr 0.369
## 4 medi_bun_t_var_vaccine_live_subq 0.325
## 5 medi_bun_t_hepa 0.301
## 6 faci_bun_t_office 0.247
## 7 faci_bun_t_pat 0.246
## 8 faci_bun_t_dept 0.204
## 9 faci_bun_t_emergency 0.201
## 10 medi_bun_t_tdap 0.182
## # ... with 219 more rowsmmr
flu %>% get_tag_density_information("medi_bun_t_mmr") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ medi_bun_t_mmr"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 19.5## $dist_plots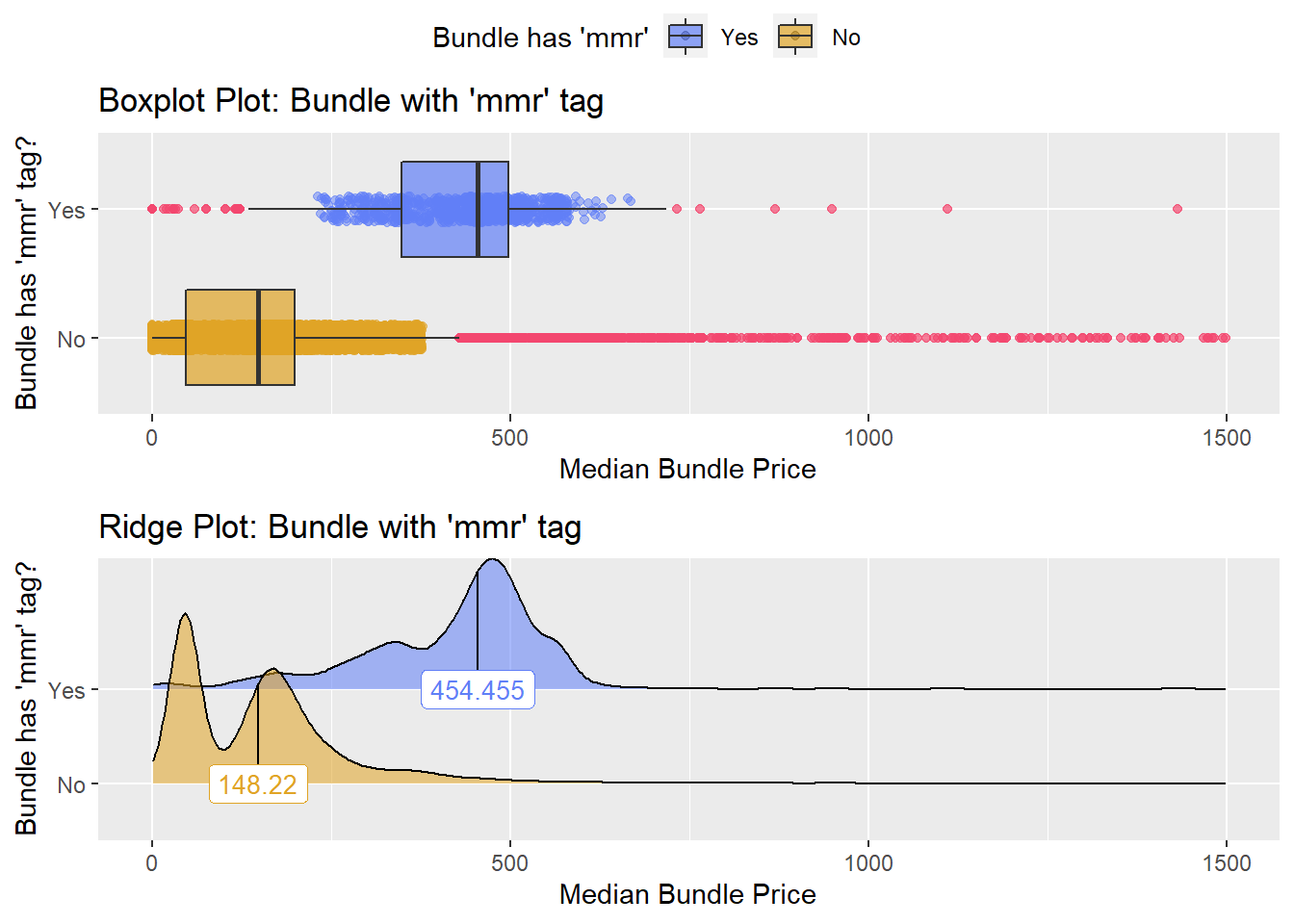
##
## $stat_tables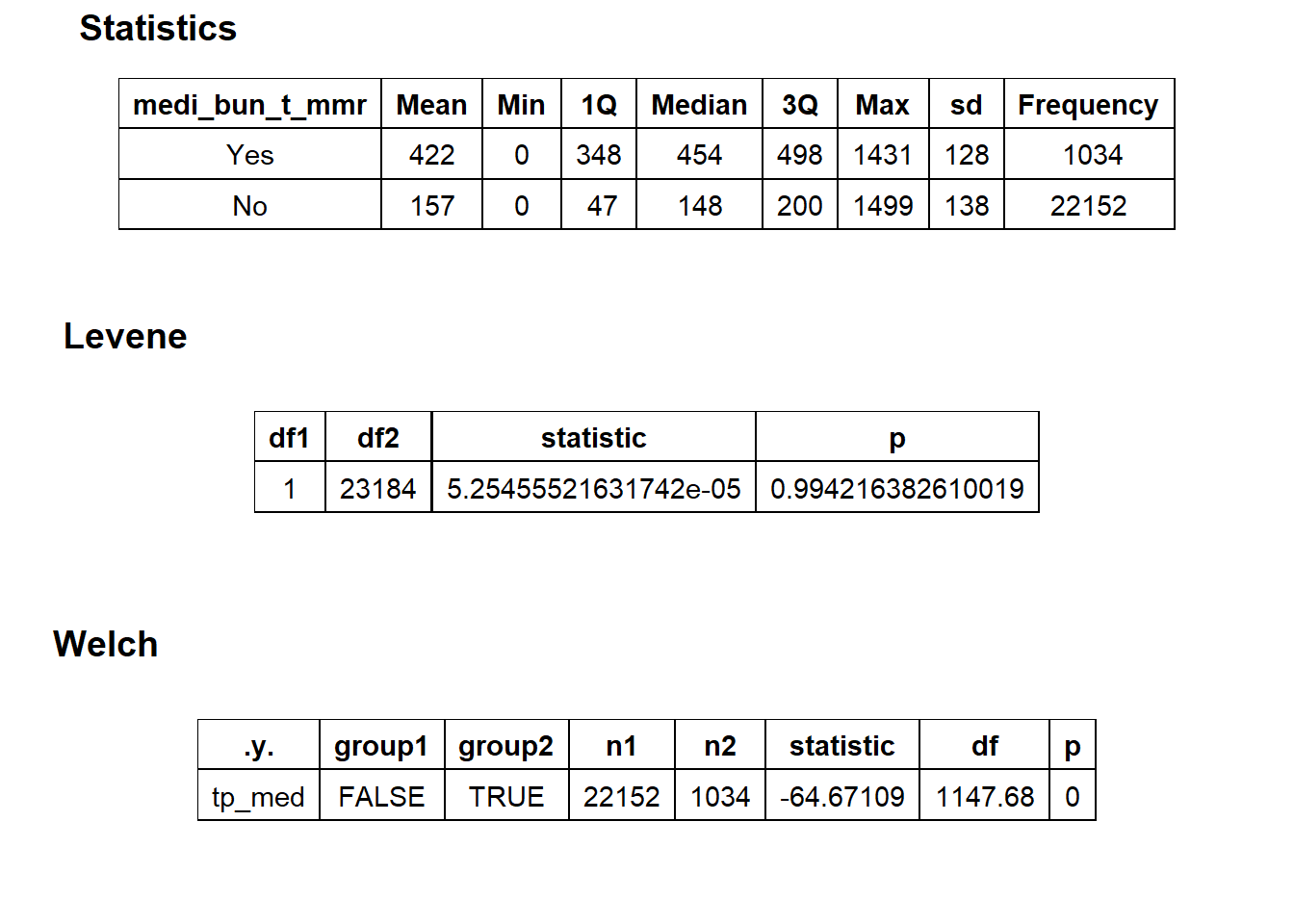
flu_w_mmr <- flu[medi_bun_t_mmr == T]
flu <- flu[medi_bun_t_mmr==F]var_vaccine_live_subq
flu %>% get_tag_density_information("medi_bun_t_var_vaccine_live_subq") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ medi_bun_t_var_vaccine_live_subq"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 22.7## $dist_plots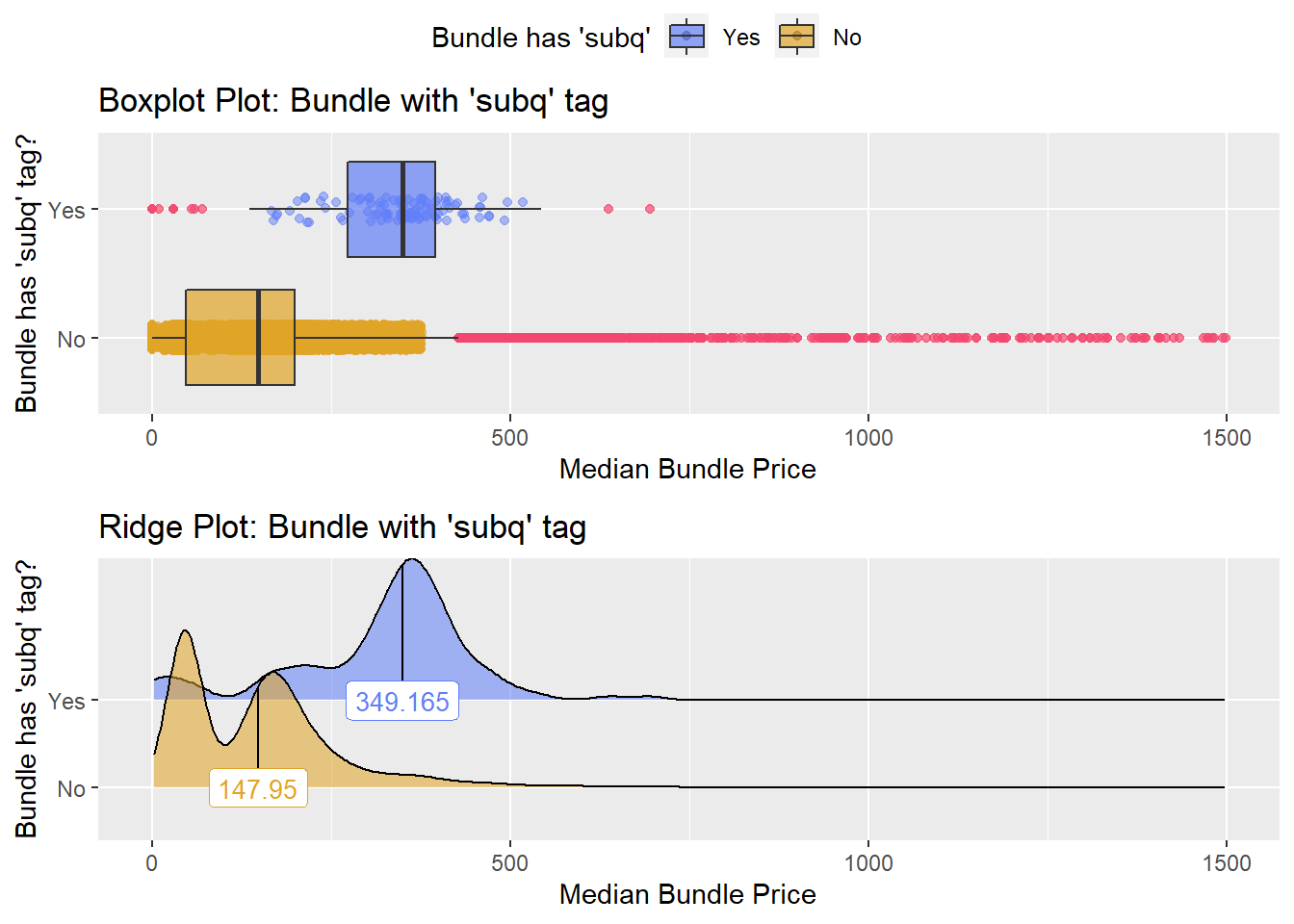
##
## $stat_tables
flu_w_vaccine_live_subq<- flu[medi_bun_t_var_vaccine_live_subq == T]
flu <- flu[medi_bun_t_var_vaccine_live_subq==F]hepa
flu %>% get_tag_density_information("medi_bun_t_hepa") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ medi_bun_t_hepa"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 9.98## $dist_plots
##
## $stat_tables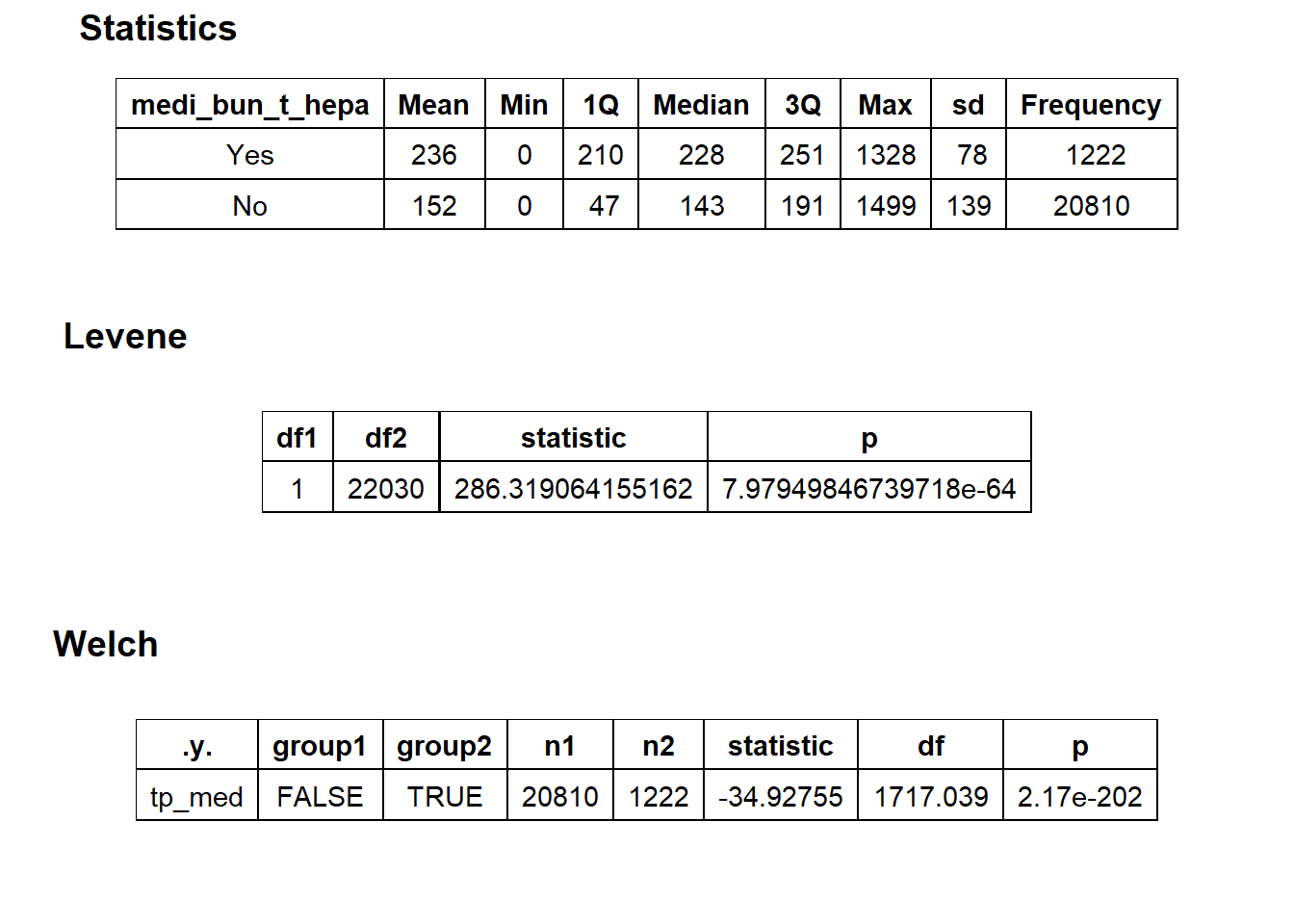
flu_w_hepa <- flu[medi_bun_t_hepa == T]
flu <- flu[medi_bun_t_hepa==F]tdap
flu %>% get_tag_density_information("medi_bun_t_tdap") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ medi_bun_t_tdap"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 16.9## $dist_plots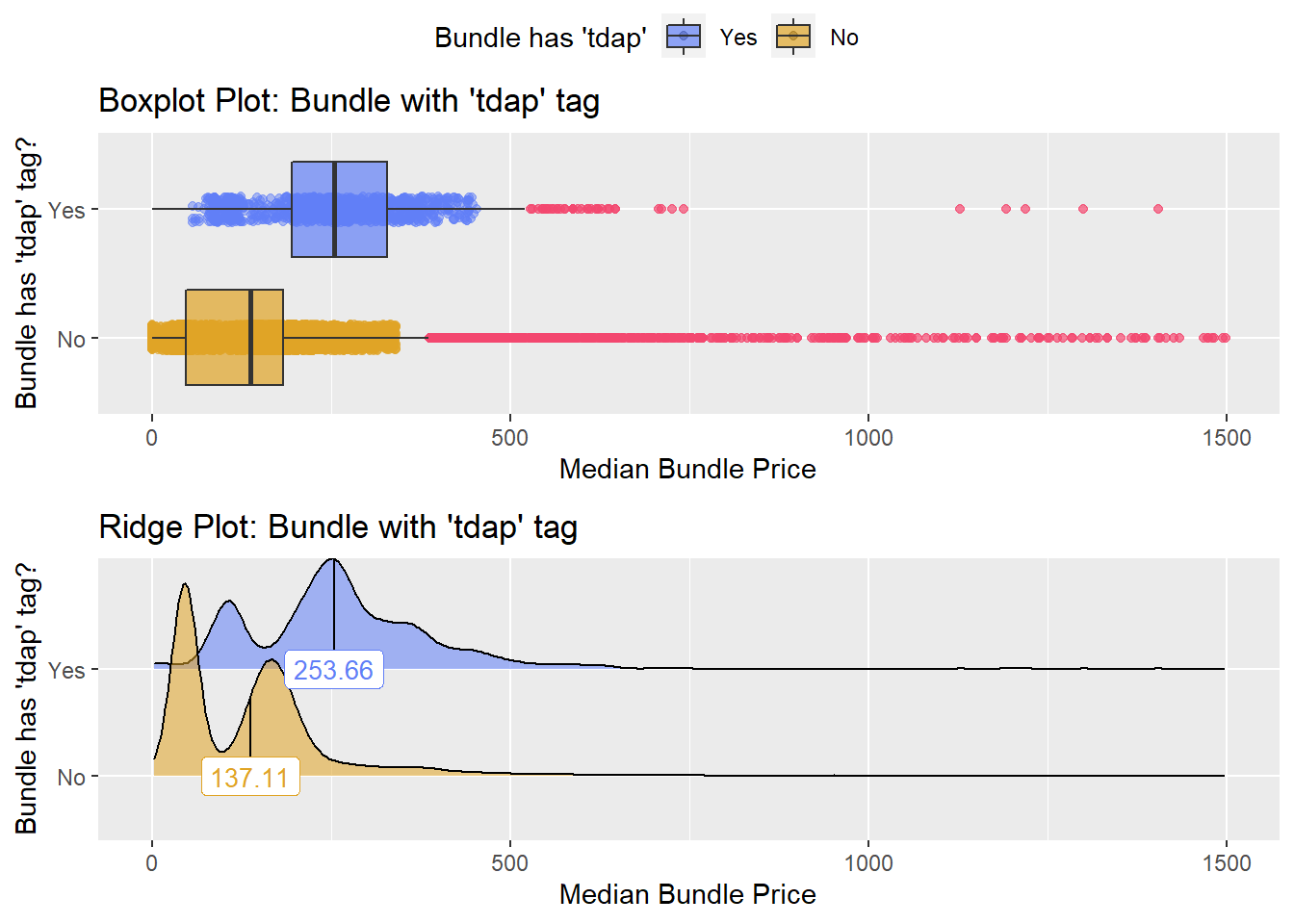
##
## $stat_tables
flu_w_tdap <- flu[medi_bun_t_tdap == T]
flu <- flu[medi_bun_t_tdap==F]dna
flu %>% get_tag_density_information("path_bun_t_dna") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ path_bun_t_dna"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 20.6## $dist_plots
##
## $stat_tables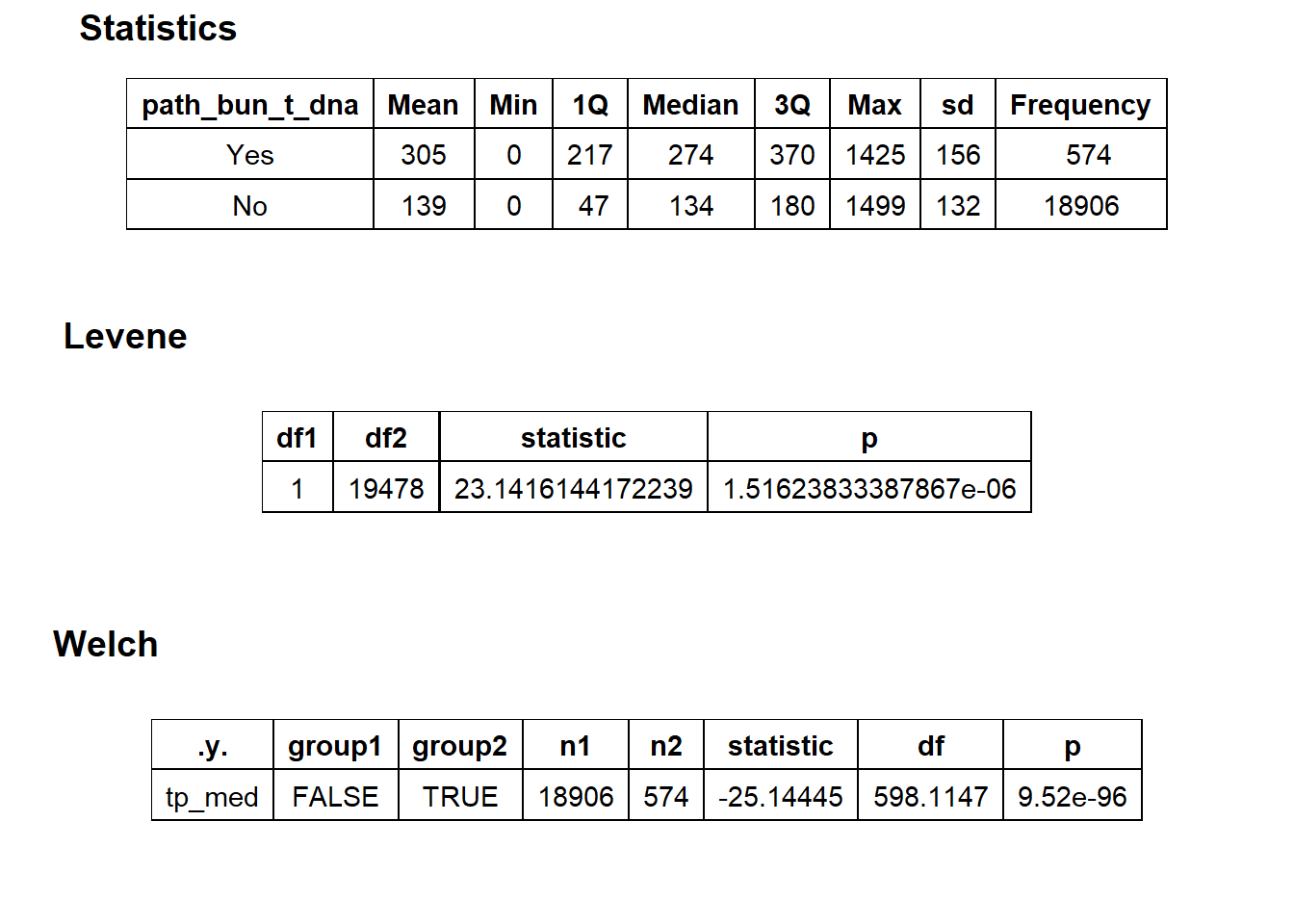
flu_w_dna <- flu[path_bun_t_dna == T]
flu <- flu[path_bun_t_dna == F]##cytopath
flu %>% get_tag_density_information("path_bun_t_cytopath") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ path_bun_t_cytopath"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 18.3## $dist_plots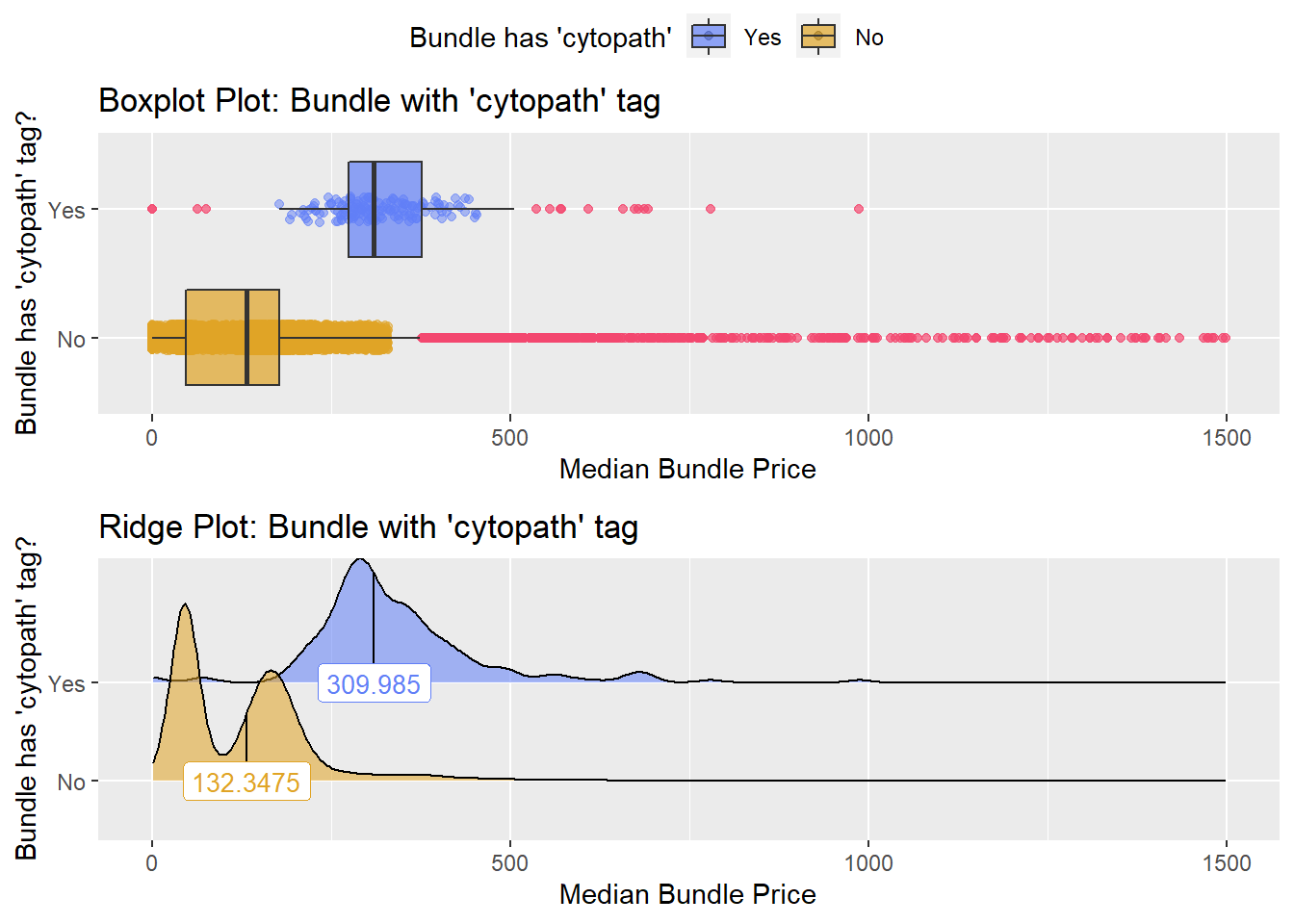
##
## $stat_tables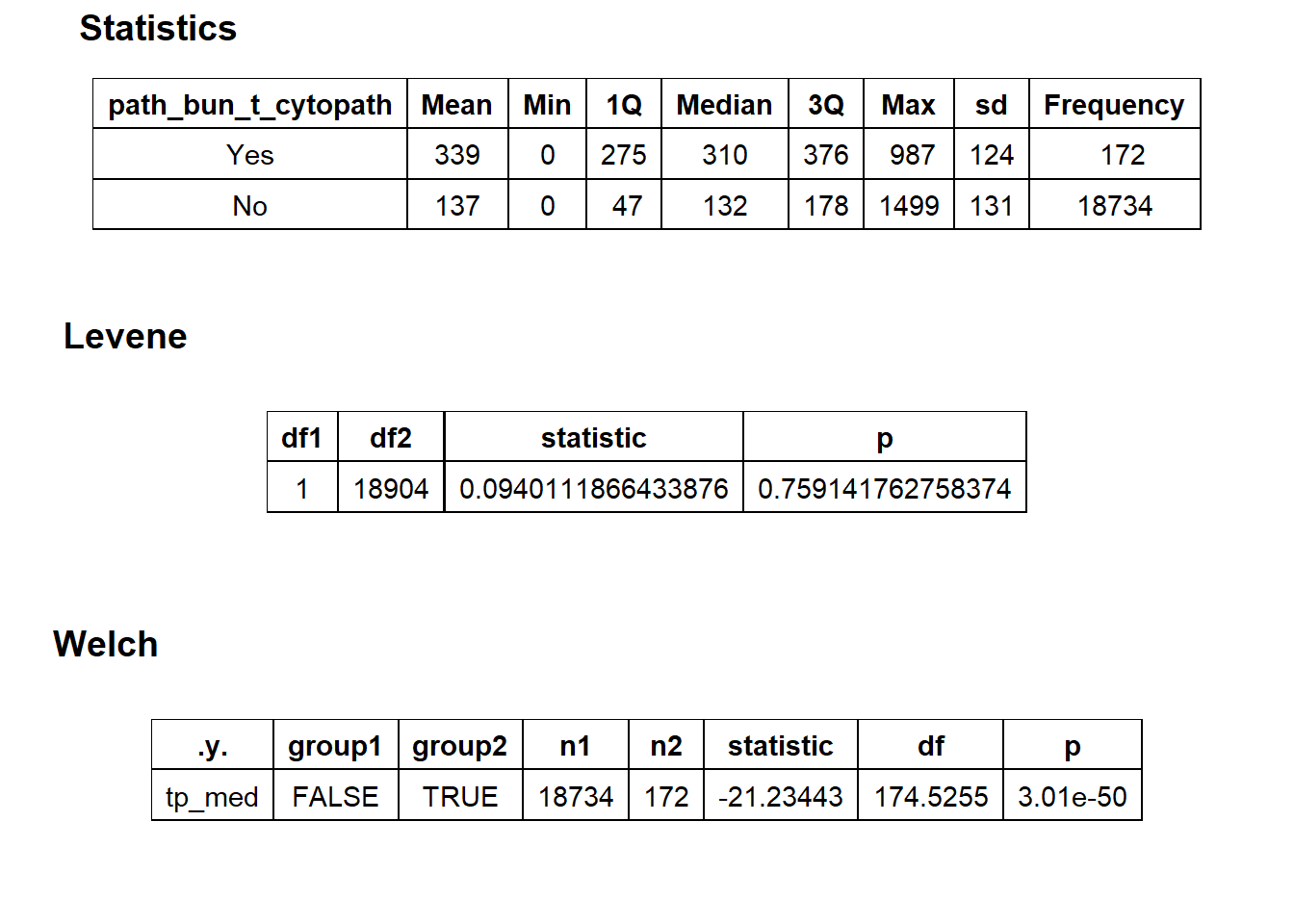
flu_w_cytopath <- flu[path_bun_t_cytopath == T]
flu <- flu[path_bun_t_cytopath == F]##hzv medi_bun_t_hzv
flu %>% get_tag_density_information("medi_bun_t_hzv") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ medi_bun_t_hzv"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 26.8## $dist_plots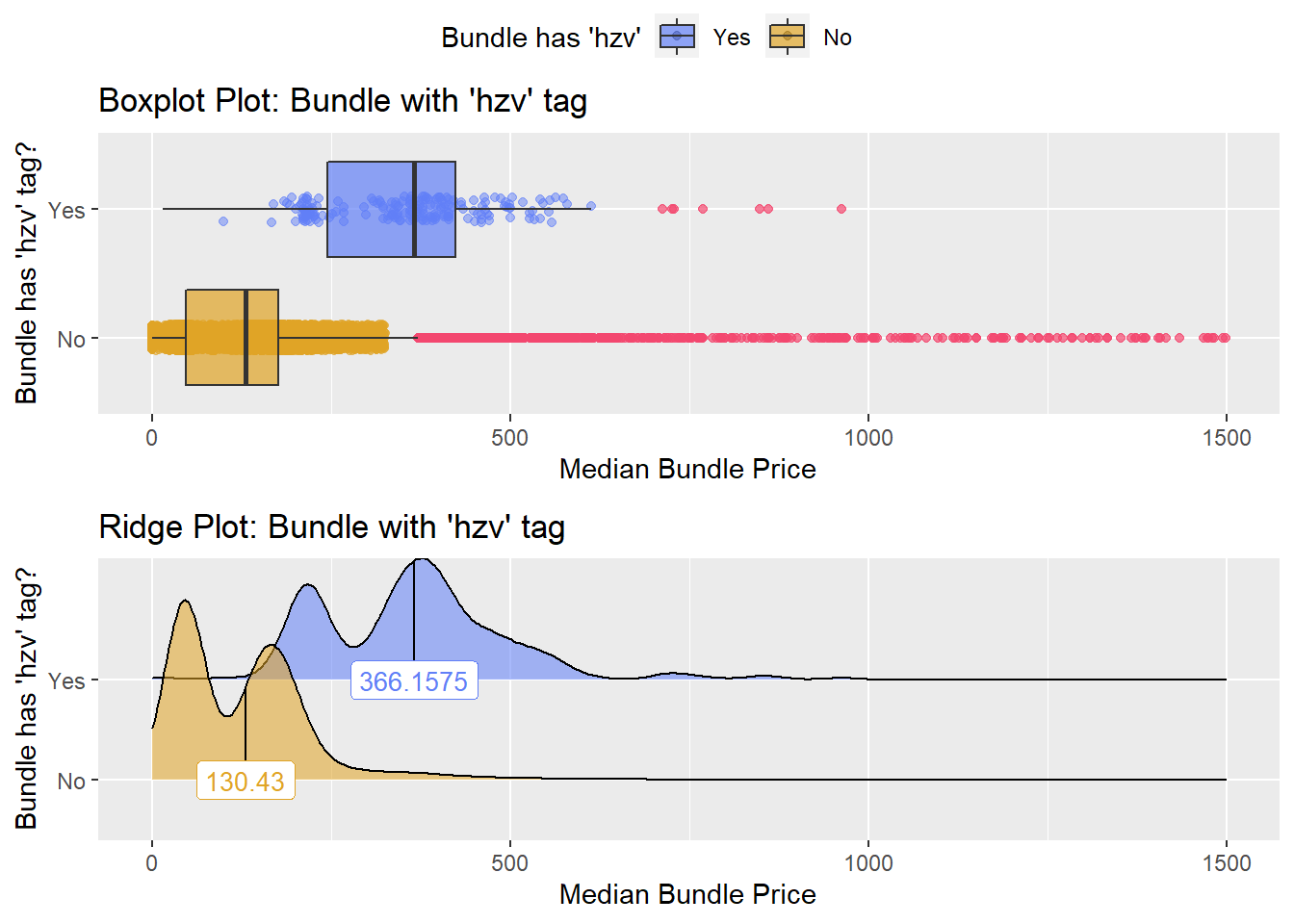
##
## $stat_tables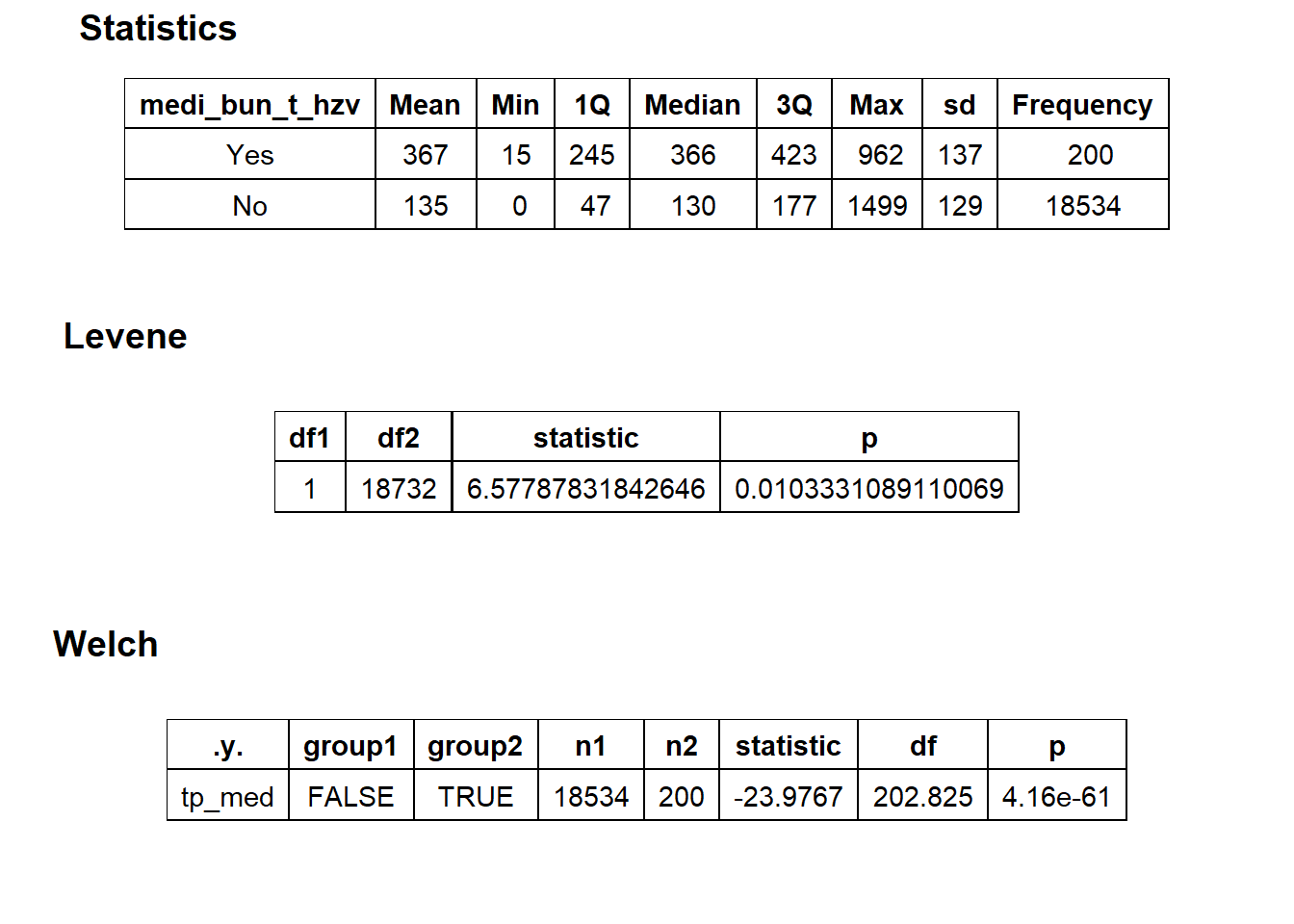
flu_w_hzv <- flu[medi_bun_t_hzv == T]
flu <- flu[medi_bun_t_hzv == F]gonorrhea
path_bun_t_gonorr
#flu %>% get_tag_density_information("path_bun_t_gonorr") %>% print()flu_w_gonorr <- flu[path_bun_t_gonorr == T]
flu <- flu[path_bun_t_gonorr == F]meningitis b
medi_bun_t_menb
flu %>% get_tag_density_information("medi_bun_t_menb") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ medi_bun_t_menb"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 13.3## $dist_plots
##
## $stat_tables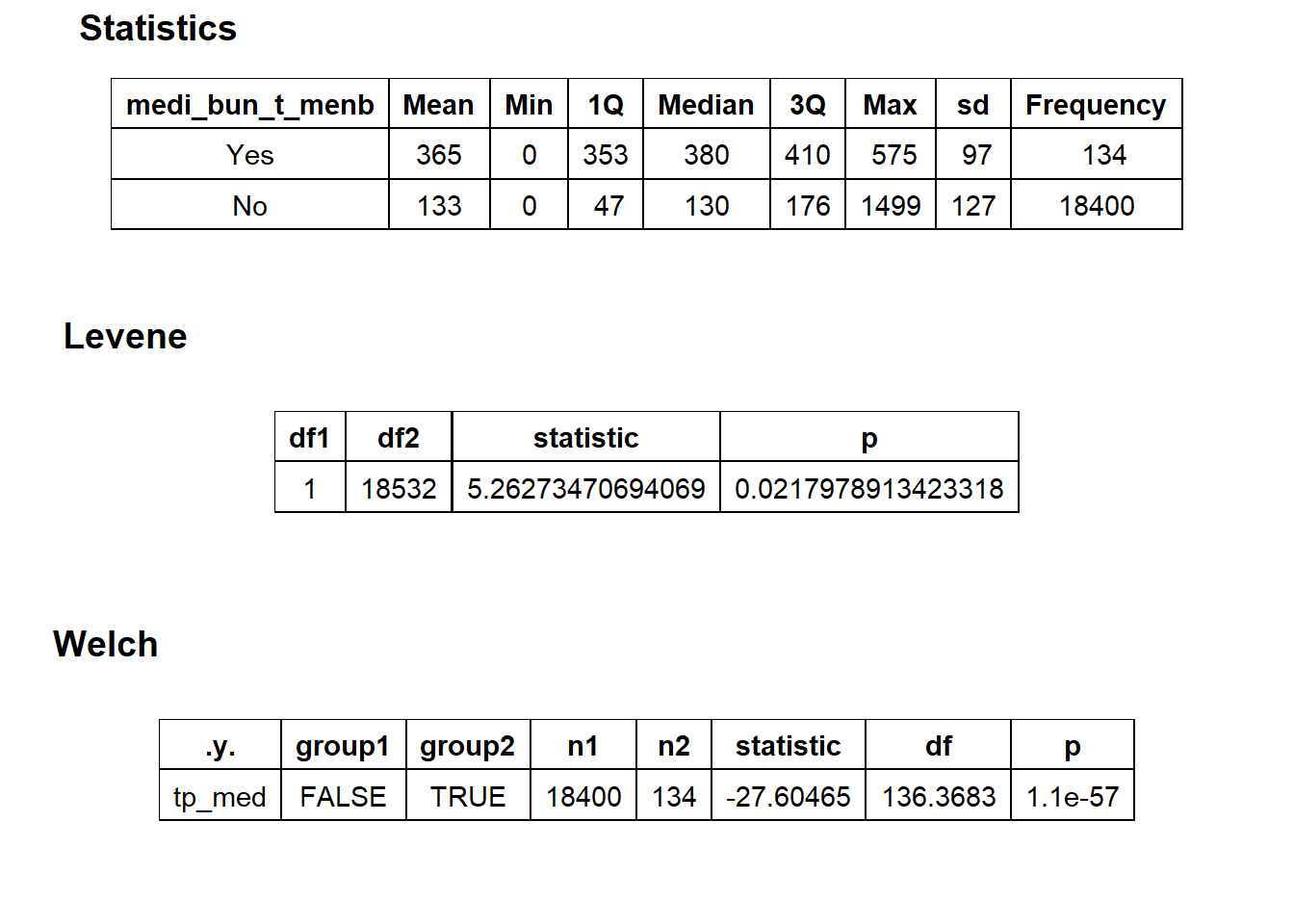
flu_w_menb <- flu[medi_bun_t_menb == T]
flu <- flu[medi_bun_t_menb == F]urinalysis
path_bun_t_urinalysis
flu %>% get_tag_density_information("path_bun_t_urinalysis") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ path_bun_t_urinalysis"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 14.5## $dist_plots
##
## $stat_tables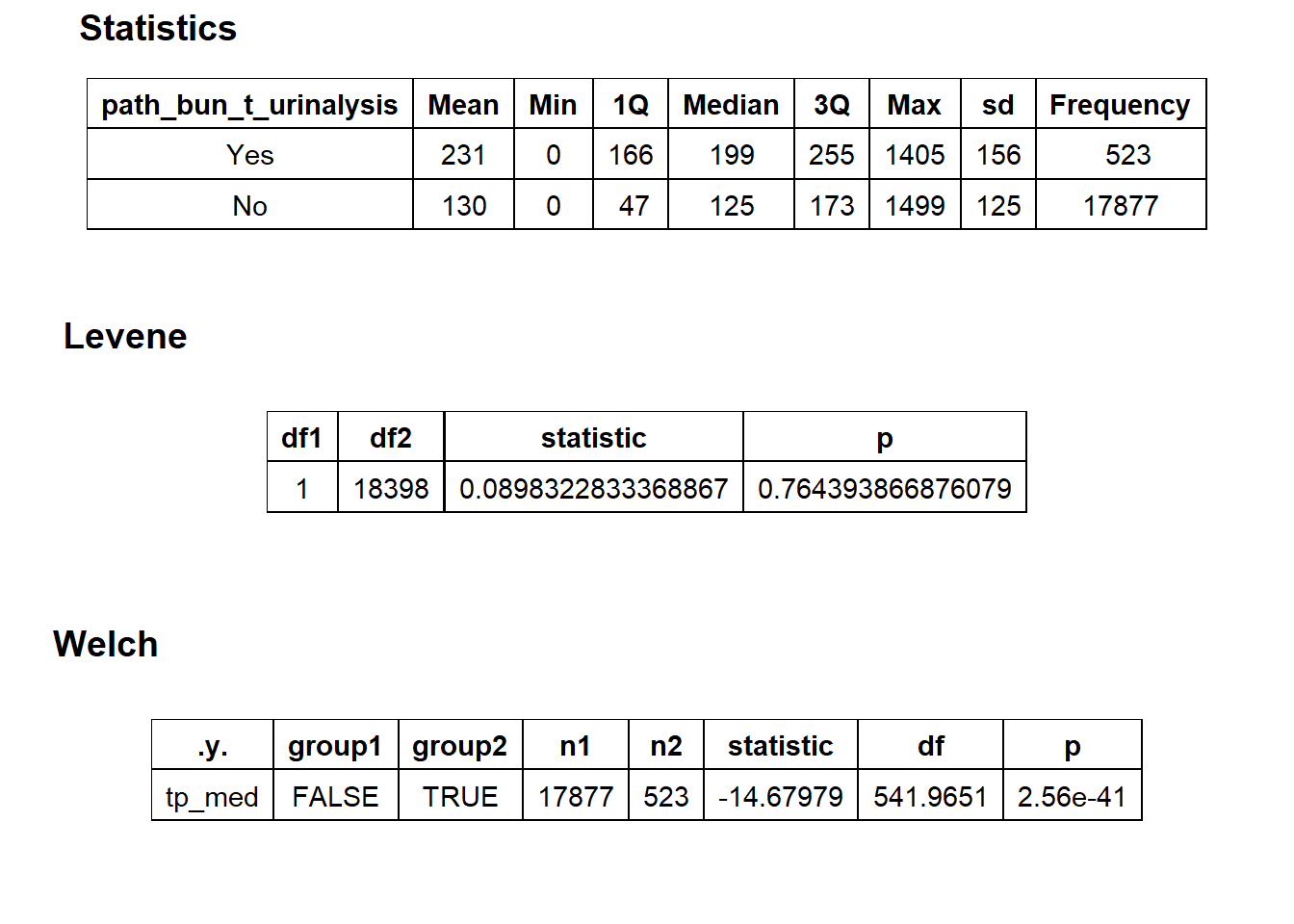
flu_w_urinalysis <- flu[path_bun_t_urinalysis == T]
flu <- flu[path_bun_t_urinalysis == F]flu %>% plot_med_density() %>% print()## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.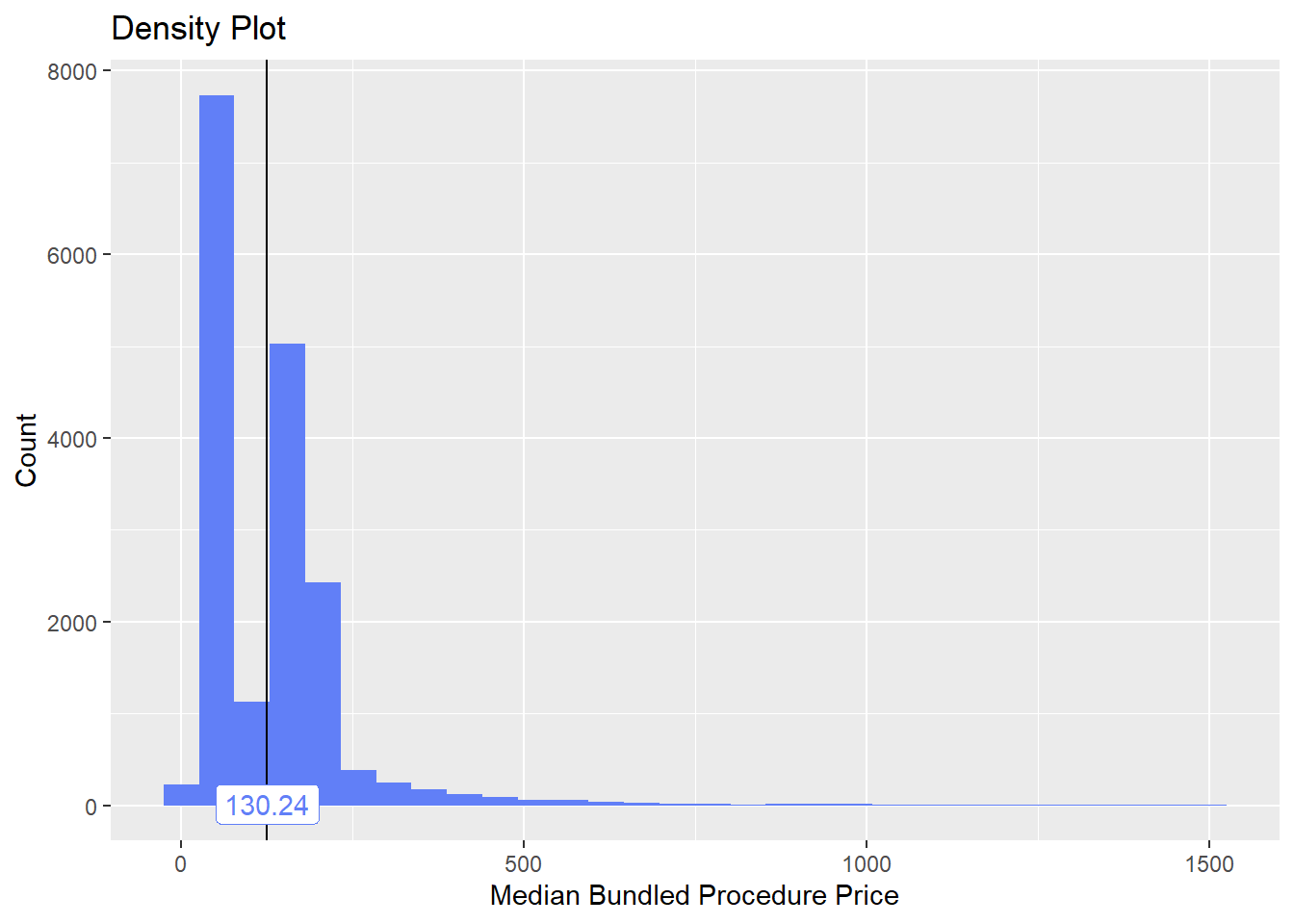
flu %>% get_tag_cor() %>% print()## Warning in stats::cor(cor_data): the standard deviation is zero## # A tibble: 185 x 2
## name correlation
## <chr> <dbl>
## 1 faci_bun_t_visit 0.537
## 2 faci_bun_t_est 0.487
## 3 faci_bun_t_pat 0.382
## 4 faci_bun_t_office 0.377
## 5 medi_bun_t_test 0.264
## 6 medi_bun_t_evaluat 0.245
## 7 faci_bun_t_dept 0.212
## 8 medi_bun_t_add-on 0.211
## 9 faci_bun_t_emergency 0.207
## 10 medi_bun_t_wheezing 0.205
## # ... with 175 more rowsflu_with_faci <- flu[faci_bun_sum_med > 0]
flu <- flu[faci_bun_sum_med==0 & cnt > 20 & tp_med > 0]flu shot only
flu %>% plot_med_density() %>% print()## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.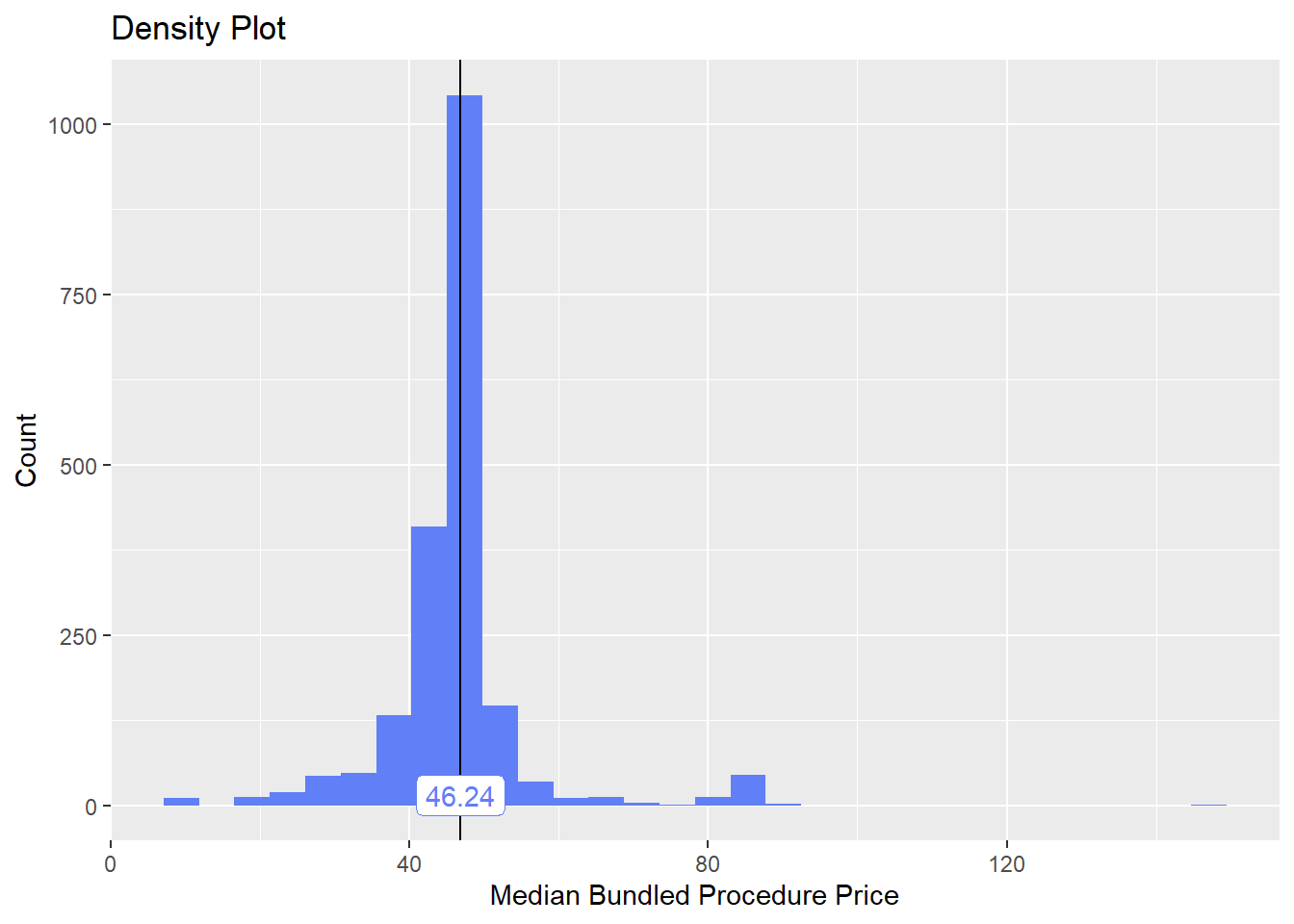
flu %>% get_tag_cor() %>% print()## Warning in stats::cor(cor_data): the standard deviation is zero## # A tibble: 20 x 2
## name correlation
## <chr> <dbl>
## 1 medi_bun_t_riv4 0.711
## 2 medi_bun_t_iiv 0.600
## 3 medi_bun_t_immun 0.254
## 4 medi_bun_t_flu 0.245
## 5 medi_bun_t_influenza 0.245
## 6 medi_bun_t_virus 0.245
## 7 medi_bun_t_ppsv23 0.233
## 8 medi_bun_t_splt 0.199
## 9 medi_bun_t_immunization 0.193
## 10 medi_bun_t_vacc 0.0504
## 11 path_bun_t_gene 0.0477
## 12 path_bun_t_general 0.0477
## 13 path_bun_t_health 0.0477
## 14 path_bun_t_panel 0.0477
## 15 medi_bun_t_immunotherapy 0.0393
## 16 medi_bun_t_inject 0.0393
## 17 medi_bun_t_laiv4 0.0288
## 18 medi_bun_t_immune 0.0283
## 19 medi_bun_t_oral 0.0283
## 20 path_bun_t_test 0.0239flu <- flu[,`:=`(
surg_sp_name_clean = surg_sp_npi %>% map_chr(get_npi_standard_name),
surg_bp_name_clean = surg_bp_npi %>% map_chr(get_npi_standard_name),
medi_sp_name_clean = medi_sp_npi %>% map_chr(get_npi_standard_name),
medi_bp_name_clean = medi_bp_npi %>% map_chr(get_npi_standard_name),
radi_sp_name_clean = radi_sp_npi %>% map_chr(get_npi_standard_name),
radi_bp_name_clean = radi_bp_npi %>% map_chr(get_npi_standard_name),
path_sp_name_clean = path_sp_npi %>% map_chr(get_npi_standard_name),
path_bp_name_clean = path_bp_npi %>% map_chr(get_npi_standard_name),
anes_sp_name_clean = anes_sp_npi %>% map_chr(get_npi_standard_name),
anes_bp_name_clean = anes_bp_npi %>% map_chr(get_npi_standard_name),
faci_sp_name_clean = faci_sp_npi %>% map_chr(get_npi_standard_name),
faci_bp_name_clean = faci_bp_npi %>% map_chr(get_npi_standard_name)
)]fix the npi subset to be much more broad
flu_btbv4 <- flu %>% btbv4_medi_fac_only()flu_btbv4 %>% glimpse## Rows: 65
## Columns: 10
## $ most_important_fac <chr> "MIDTOWN COMMUNITY HEALTH CENTER (OGDEN)", ...
## $ tp_med_med <dbl> 9.000, 46.550, 29.330, 22.000, 38.570, 42.76...
## $ tp_med_surg <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,...
## $ tp_med_medi <dbl> 9.000, 46.550, 29.330, 22.000, 38.570, 42.76...
## $ tp_med_radi <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,...
## $ tp_med_path <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,...
## $ tp_med_anes <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,...
## $ tp_med_faci <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,...
## $ tp_cnt_cnt <int> 816, 151343, 4996, 450, 250, 1682, 1256, 313...
## $ most_important_fac_npi <chr> "1548267420", "0", "1588656870", "1881001378...flu_bq <- flu_btbv4[,`:=`(procedure_type=8, procedure_modifier="Standard")]flu_bq <- flu_bq[,.(
most_important_fac ,
most_important_fac_npi,
procedure_type,
procedure_modifier,
tp_med_med,
tp_med_surg,
tp_med_medi,
tp_med_path,
tp_med_radi,
tp_med_anes,
tp_med_faci,
ingest_date = Sys.Date()
)]bq_table_upload(x=procedure_fac_only_table, values= flu_bq, create_disposition='CREATE_IF_NEEDED', write_disposition='WRITE_APPEND')flu with a facility / visit charge
flu_with_faci %>% plot_med_density() %>% print()## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.
flu_with_faci %>% get_tag_cor() %>% print()## Warning in stats::cor(cor_data): the standard deviation is zero## # A tibble: 177 x 2
## name correlation
## <chr> <dbl>
## 1 faci_bun_t_dept 0.273
## 2 medi_bun_t_evaluat 0.272
## 3 faci_bun_t_emergency 0.264
## 4 medi_bun_t_test 0.25
## 5 medi_bun_t_wheezing 0.230
## 6 medi_bun_t_add-on 0.220
## 7 medi_bun_t_visit 0.206
## 8 medi_bun_t_clinic 0.201
## 9 medi_bun_t_outpt 0.201
## 10 medi_bun_t_stud 0.194
## # ... with 167 more rowsemergency department
flu_with_faci %>% get_tag_density_information("faci_bun_t_emergency") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ faci_bun_t_emergency"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 105## $dist_plots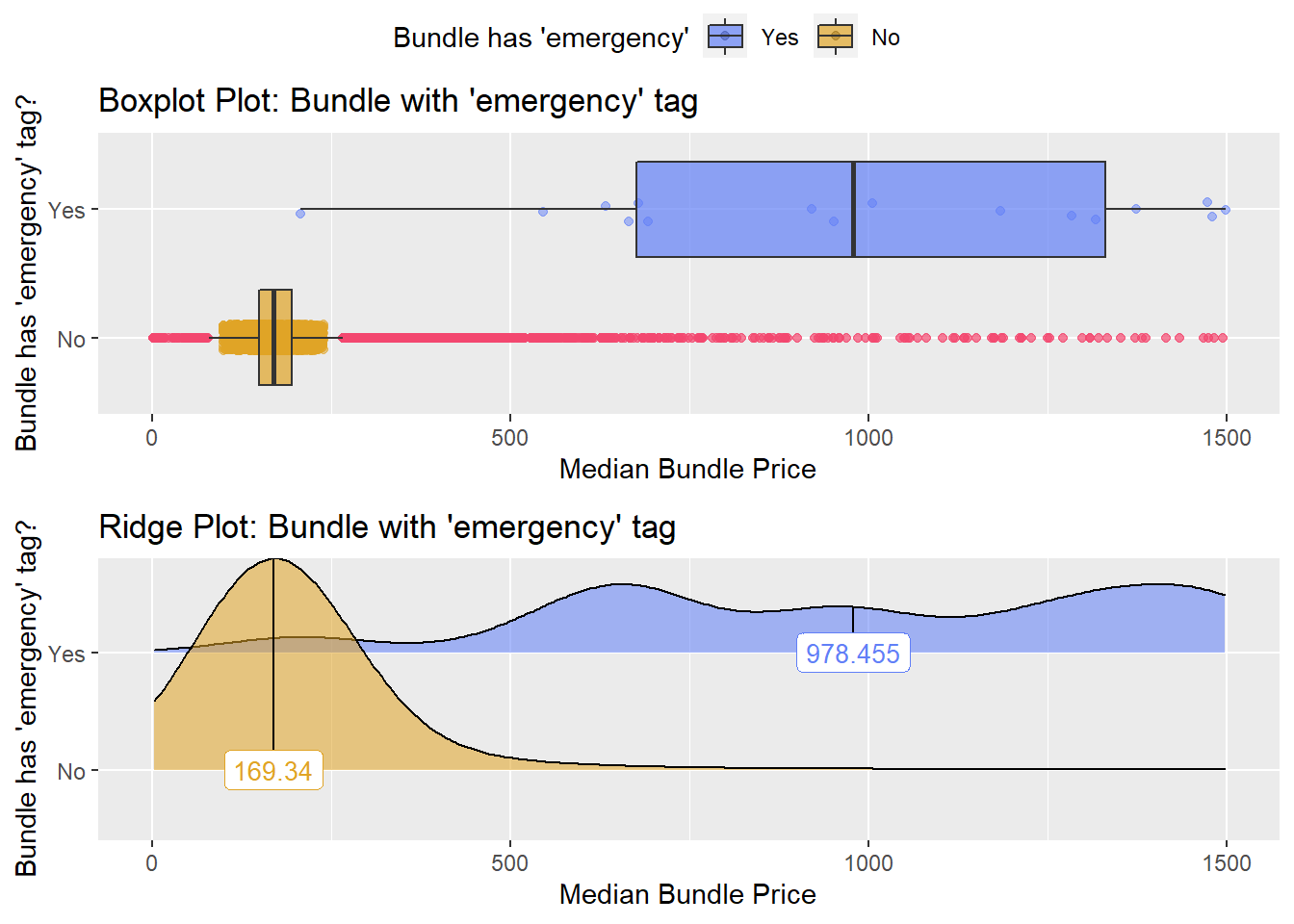
##
## $stat_tables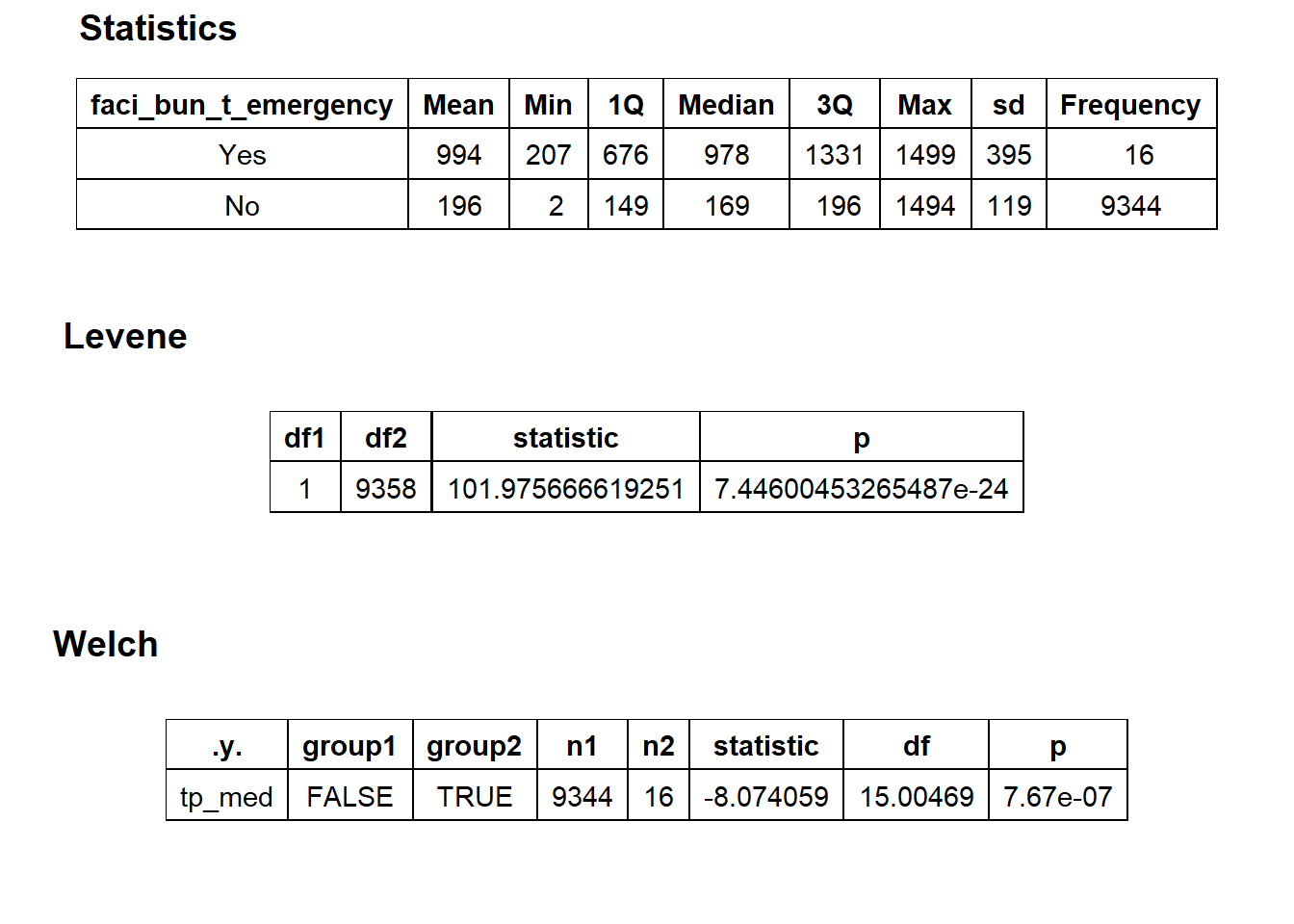
# flu_with_faci_w_emergency <- flu_with_faci[faci_bun_t_emergency==T]
flu_with_faci <- flu_with_faci[faci_bun_t_emergency==F]medi_bun_t_wheezing
flu_with_faci %>% get_tag_density_information("medi_bun_t_wheezing") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ medi_bun_t_wheezing"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 73.2## $dist_plots
##
## $stat_tables
flu_with_faci <- flu_with_faci[medi_bun_t_wheezing==F]flu_with_faci %>% plot_med_density() %>% print()## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.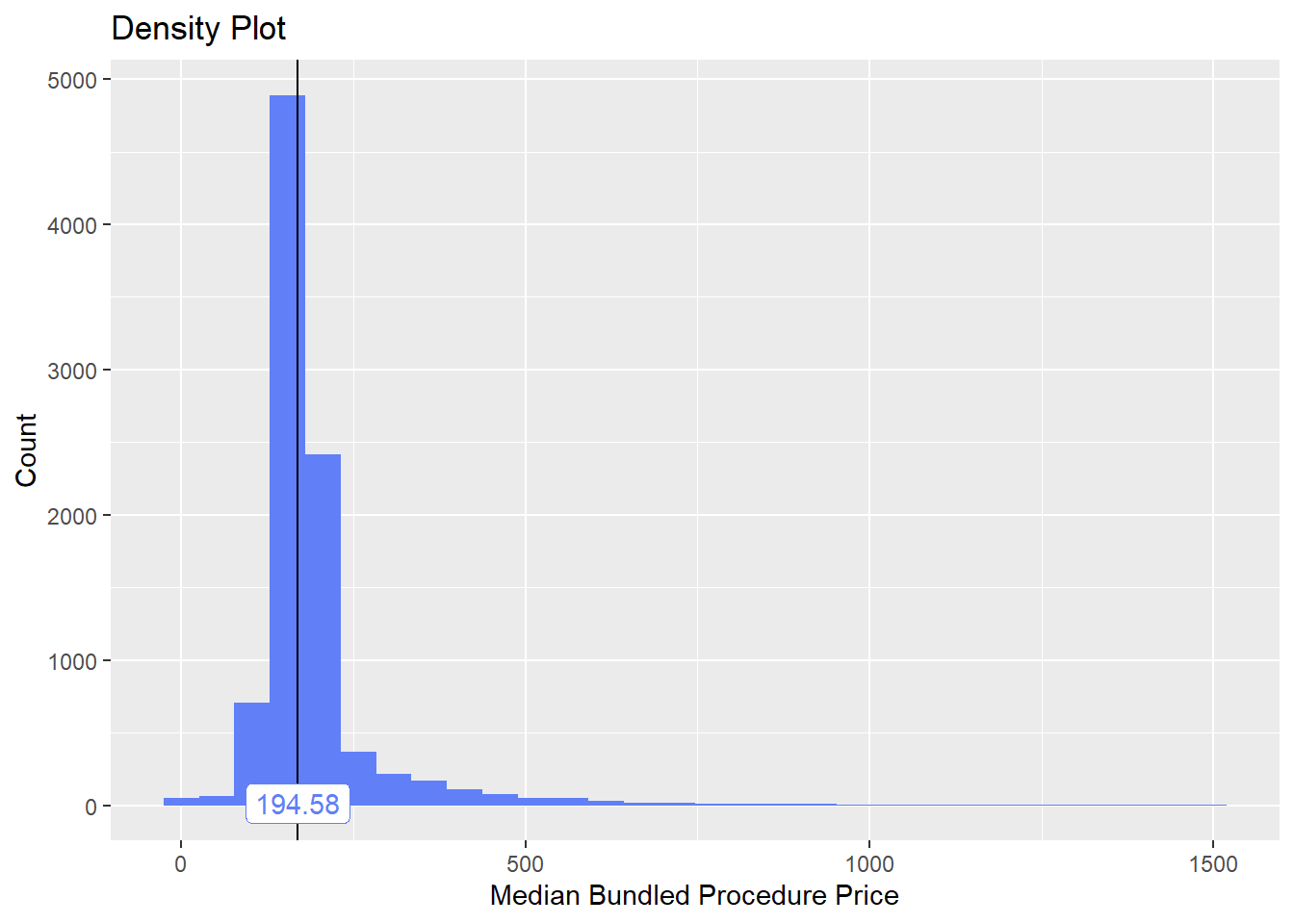
flu_with_faci %>% get_tag_cor() %>% print()## Warning in stats::cor(cor_data): the standard deviation is zero## # A tibble: 173 x 2
## name correlation
## <chr> <dbl>
## 1 medi_bun_t_test 0.253
## 2 medi_bun_t_add-on 0.213
## 3 medi_bun_t_therapeutic 0.198
## 4 medi_bun_t_visit 0.197
## 5 medi_bun_t_stud 0.196
## 6 medi_bun_t_clinic 0.190
## 7 medi_bun_t_outpt 0.190
## 8 medi_bun_t_ppsv23 0.188
## 9 medi_bun_t_tests 0.181
## 10 path_bun_t_immuno 0.180
## # ... with 163 more rowsflu_with_faci %>% get_tag_density_information("medi_bun_t_therapeutic") %>% print()## Scale for 'y' is already present. Adding another scale for 'y', which will
## replace the existing scale.## Scale for 'x' is already present. Adding another scale for 'x', which will
## replace the existing scale.## [1] "tp_med ~ medi_bun_t_therapeutic"## Warning in leveneTest.default(y = y, group = group, ...): group coerced to
## factor.## Picking joint bandwidth of 23.1## $dist_plots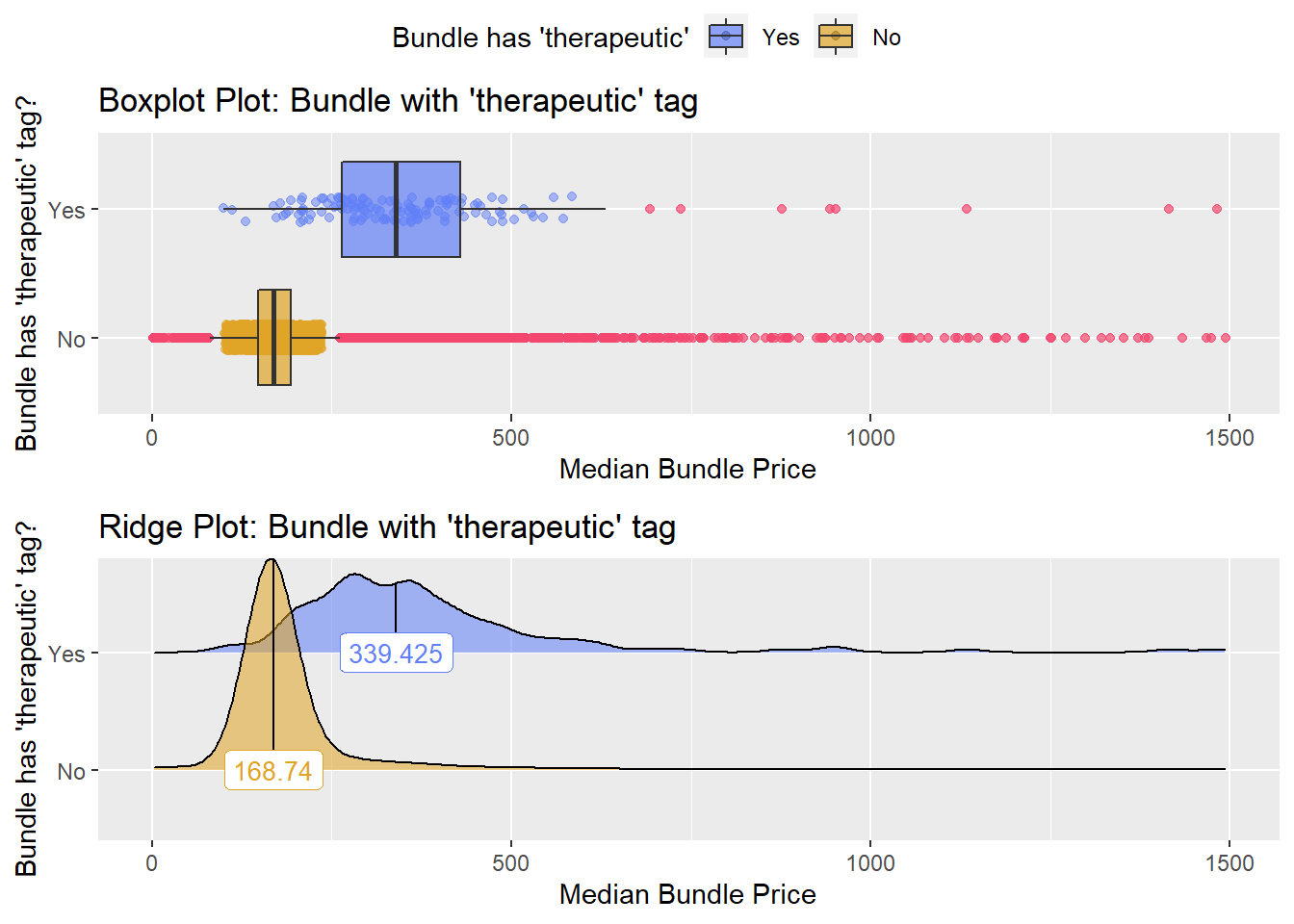
##
## $stat_tables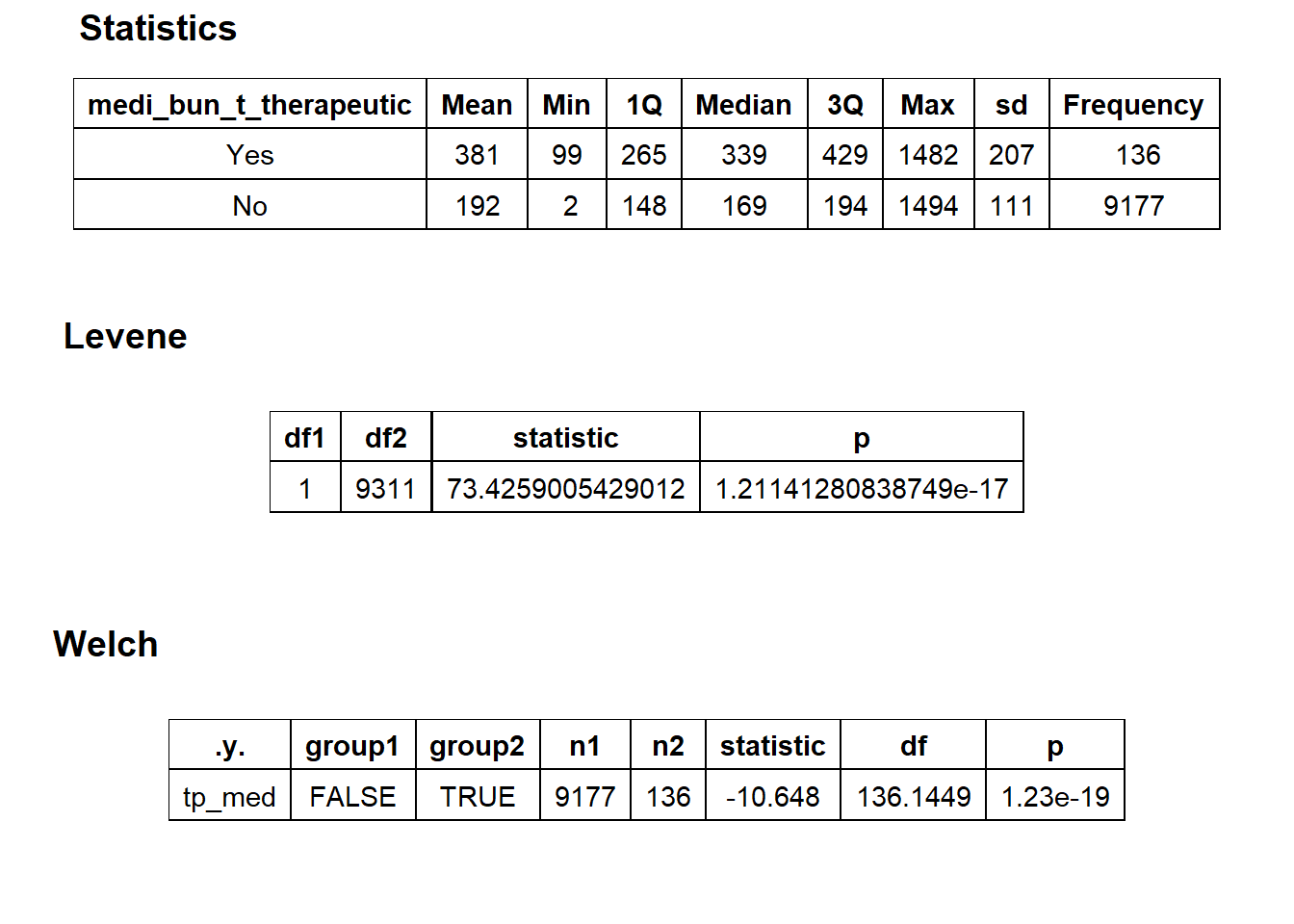
flu_with_faci <- flu_with_faci[medi_bun_t_therapeutic==F & medi_bun_t_exercise==F & medi_bun_t_manual==F & path_bun_t_culture ==F & path_bun_t_microbe==F & medi_bun_t_psytx==F & medi_bun_t_eye_exam==F & medi_bun_t_stress==F &medi_bun_t_services==F & medi_bun_t_toxin==F & medi_bun_t_tracing==F & medi_bun_t_psych==F & medi_bun_t_speech==F & medi_bun_t_reeducation==F & path_bun_t_gene==F & surg_bun_t_colonoscopy==F]flu_with_faci %>% plot_med_density() %>% print()## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.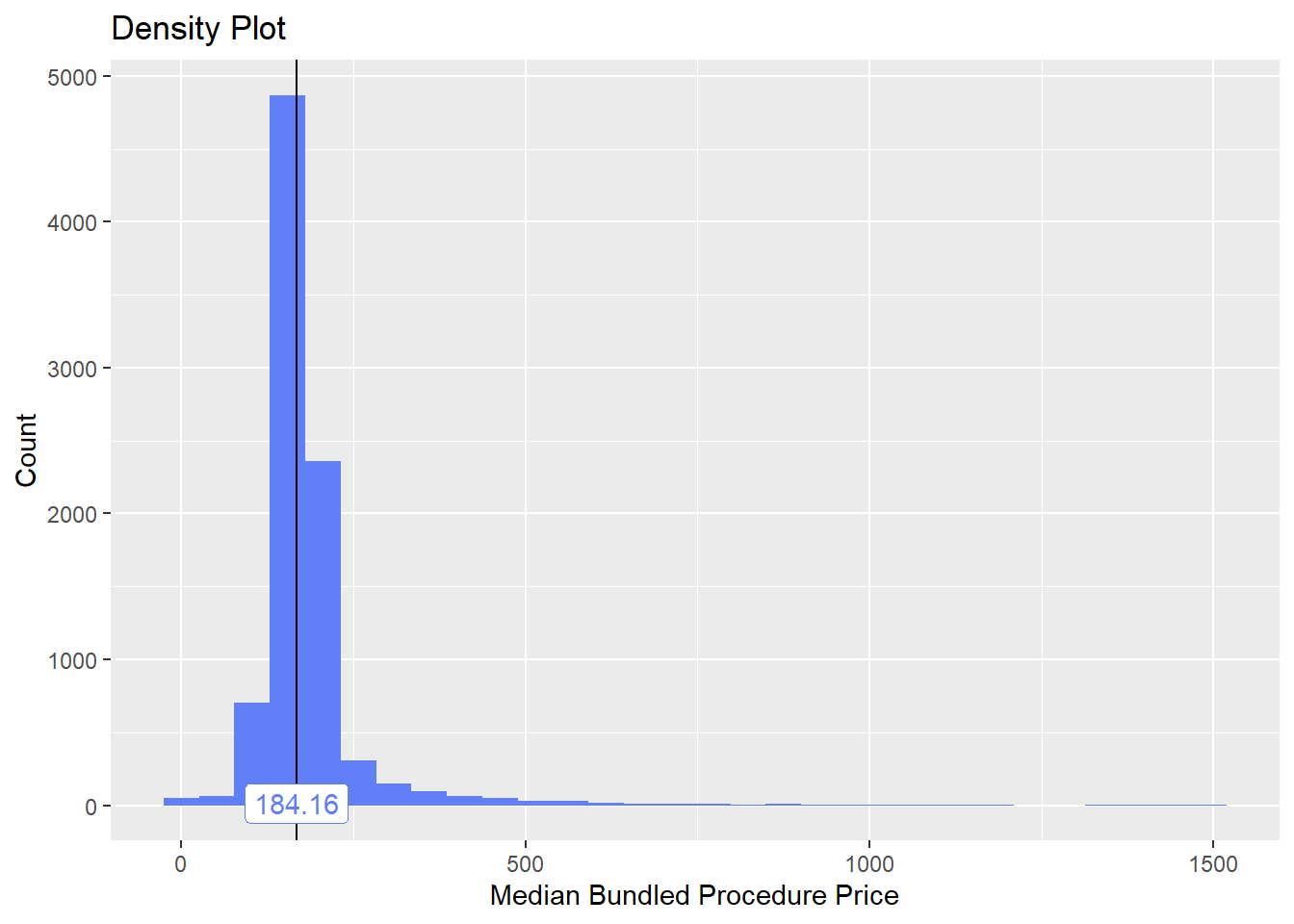
flu_with_faci %>% get_tag_cor() %>% print()## Warning in stats::cor(cor_data): the standard deviation is zero## # A tibble: 130 x 2
## name correlation
## <chr> <dbl>
## 1 medi_bun_t_stud 0.248
## 2 medi_bun_t_test 0.228
## 3 medi_bun_t_ppsv23 0.226
## 4 medi_bun_t_add-on 0.210
## 5 medi_bun_t_tests 0.169
## 6 medi_bun_t_inject 0.151
## 7 medi_bun_t_visit 0.148
## 8 faci_bun_t_est 0.145
## 9 medi_bun_t_flu 0.138
## 10 medi_bun_t_influenza 0.138
## # ... with 120 more rowsflu_with_faci <- flu_with_faci[medi_bun_t_stud==F & medi_bun_t_test==F & `medi_bun_t_add-on`==F & radi_bun_t_abdom==F & medi_bun_t_swallow==F & path_bun_t_pregnancy==F ]flu_with_faci$tp_med %>% summary() ## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 1.5 146.6 166.0 178.1 190.7 1494.3flu_with_faci %>% get_tag_cor() %>% print()## Warning in stats::cor(cor_data): the standard deviation is zero## # A tibble: 108 x 2
## name correlation
## <chr> <dbl>
## 1 medi_bun_t_ppsv23 0.236
## 2 medi_bun_t_inject 0.141
## 3 medi_bun_t_visit 0.132
## 4 faci_bun_t_est 0.125
## 5 medi_bun_t_clinic 0.107
## 6 medi_bun_t_outpt 0.107
## 7 medi_bun_t_asses 0.105
## 8 medi_bun_t_riv4 0.0923
## 9 medi_bun_t_virus 0.091
## 10 medi_bun_t_flu 0.0907
## # ... with 98 more rowsflu_with_faci <- flu_with_faci[,`:=`(
surg_sp_name_clean = surg_sp_npi %>% map_chr(get_npi_standard_name),
surg_bp_name_clean = surg_bp_npi %>% map_chr(get_npi_standard_name),
medi_sp_name_clean = medi_sp_npi %>% map_chr(get_npi_standard_name),
medi_bp_name_clean = medi_bp_npi %>% map_chr(get_npi_standard_name),
radi_sp_name_clean = radi_sp_npi %>% map_chr(get_npi_standard_name),
radi_bp_name_clean = radi_bp_npi %>% map_chr(get_npi_standard_name),
path_sp_name_clean = path_sp_npi %>% map_chr(get_npi_standard_name),
path_bp_name_clean = path_bp_npi %>% map_chr(get_npi_standard_name),
anes_sp_name_clean = anes_sp_npi %>% map_chr(get_npi_standard_name),
anes_bp_name_clean = anes_bp_npi %>% map_chr(get_npi_standard_name),
faci_sp_name_clean = faci_sp_npi %>% map_chr(get_npi_standard_name),
faci_bp_name_clean = faci_bp_npi %>% map_chr(get_npi_standard_name)
)]flu_with_faci_btbv4 <- flu_with_faci %>% btbv4_medi_fac_only()flu_with_faci_btbv4 %>% glimpse## Rows: 164
## Columns: 10
## $ most_important_fac <chr> "UNIVERSITY OF UTAH (UNIVERSITY HEALTH CARE ...
## $ tp_med_med <dbl> 314.4300, 165.2700, 224.4700, 246.7625, 160....
## $ tp_med_surg <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,...
## $ tp_med_medi <dbl> 46.8600, 46.8100, 58.9700, 78.3650, 47.2200,...
## $ tp_med_radi <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,...
## $ tp_med_path <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,...
## $ tp_med_anes <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,...
## $ tp_med_faci <dbl> 188.1150, 118.2000, 162.1800, 168.8700, 115....
## $ tp_cnt_cnt <int> 1614, 62256, 756, 951, 1503, 995, 486, 141, ...
## $ most_important_fac_npi <chr> "1588656870", "0", "1447239355", "1184657041...flu_with_faci_bq <- flu_with_faci_btbv4[,`:=`(procedure_type=8, procedure_modifier="with Doctor Visit")]flu_with_faci_bq <- flu_with_faci_bq[,.(
most_important_fac ,
most_important_fac_npi,
procedure_type,
procedure_modifier,
tp_med_med,
tp_med_surg,
tp_med_medi,
tp_med_path,
tp_med_radi,
tp_med_anes,
tp_med_faci,
ingest_date = Sys.Date()
)]bq_table_upload(x=procedure_fac_only_table, values= flu_with_faci_bq, create_disposition='CREATE_IF_NEEDED', write_disposition='WRITE_APPEND')